

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Congenital diaphragmatic hernia (CDH), a defect of the diaphragm which allows abdominal organs to herniate into the thoracic cavity, is serious and complex condition with high perinatal mortality. CDH results from failure of the pleuroperitoneal membrane to fuse in the first trimester and impacts on developing fetal lung (1). The incidence of CDH is approximately one in 2,200 to 5,000 pregnancies (2). The perinatal mortality is up to 70% and directly related to the severity of lung hypoplasia and other major combined anomalies (2, 3).
Additional structural anomalies such as congenital heart defects, neural tube defects, hydronephrosis, renal agenesis, and anencephaly are present in 25% to 57% of all cases of CDH (4-6). The defect has also been reported in association with multiple syndromes (2, 7, 8). Chromosomal abnormalities are detected in 9.5% to 34% of prenatally diagnosed cases. The most common diagnoses include trisomy 21, 18 and 13 (2, 9, 10). Other complex karyotypic abnormalities including translocations, deletions, and marker chromosomes have been reported (11). However, CDHs with Y-autosome translocation are extremely rare.
We present a case of isolated CDH combined with mosaic partial trisomy 1 of an unbalanced translocation between the long arm of the Y chromosome and the long arm of the chromosome 1.
CASE REPORT
A 25-yr-old nulliparous woman with unremarkable past medical history and no significant prenatal risk factor was referred to our department at 19 weeks' gestation for level II ultrasound examination, which revealed a fluid-filled structure in the thoracic cavity. Her previous medical history revealed that she had menarche at 16 yr of age and regular menstrual cycle before conceiving. She had a routine antenatal care. Maternal serum triple marker test was negative, showing 1.191 MoM (48.63 ng/mL) of α-FP, 0.368 MoM (14.37 IU/mL) of hCG, and 0.789 MoM (1.47 ng/mL) of uE3.
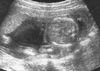
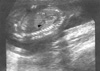
On ultrasonography a single fetus was present and fetal biometry was consistent with the patient's dates. Transabdominal ultrasonography showed a fluid-filled stomach in the left thorax with the heart displaced to the right in the axial scan (Fig. 1). Sagittal scan showed a defect in the left posterolateral diaphragm with herniation of stomach (Fig. 2). There was no associated anomaly in central nervous system, heart, both kidneys and extremities in ultrasonography. Neither polyhydramnios nor oligohydramnios was found. In order to rule out chromosomal abnormalities, we performed a cordocentesis at 20 weeks' gestation. Chromosome analysis was performed using the GTG band method (G-banding by the Trypsin-Giemsa technique). Fetal karyotype is 46,XY/46,X,-Y,+der(Y)t(Y;1)(q12;q12) (Fig. 3).
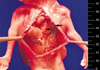
The couple was informed of the diagnosis and decided to have a termination of pregnancy at 21 weeks' gestation. A male fetus weighing 180 g was delivered vaginally. An autopsy after delivery demonstrated an isolated CDH of Bochdalek (Fig. 4). Her non-consanguineous husband was 29-yr old. Paternal karyotyping was done to assess whether the Y-autosomal translocation is transmitted from father, which subsequently revealed normal karyotype, 46, XY. Maternal karyotype was also normal.
DISCUSSION
Many authors have reported that CDH was frequently combined with additional anomalies or chromosomal defects (6, 9). Associated anomalies including congenital heart defects, hydronephrosis, renal agenesis, intestinal atresia, extralobar sequestrations, hydrocephalus, anencephaly and spina bifida are seen in 26 to 57% of all cases of CDH, but this figures rise to 95% in stillbirths. CDH has also been described as a part of syndrome such as Frey's syndrome, Beckwith-Wiedemann syndrome, Di George syndrome, Apert syndrome, Goldenhar's syndrome, Fryns syndrome and Pierre-Robin syndrome (12-15).
The incidence of chromosomal abnormalities is high, varying between 9.5% to 34% of cases in prenatally diagnosed CDH (11, 14). Trisomy 21, 18 and 13 are the most common chromosomal anomalies in fetuses with CDH (2, 10). Other complex structural chromosomal abnormalities are translocations, deletions, duplications and marker chromosomes. Clark and Fenner-Gonzales have described one case of a duplication of chromosome 1q24-q31.2, presenting with diaphragmatic hernia and other anomalies associated with Fryns syndrome (16). A chromosome 1q32-q42 deletion has been reported to be associated with diaphragmatic hernia (17). Hilfiker et al. (18) presented a case with partial reduplication of chromosome 8q (46,XY,8q+), which showed marked growth retardation, severe lung hypoplasia, left ventricular hypoplasia, and CDH, representing a potentially fatal anomaly. A chromosomal deletions or translocations of 15q21-6 with CDH have been reported, which include de novo reciprocal translocation 1:15 with tetralogy of Fallot, deletions of 15q24-ter with other malformations, balanced translocation 46,XY,t(5;15)(p15.3;q24) and deletion 15q25-q26.2 with growth retardation (19-22).
Complete or mosaic trisomy for all of chromosome 1q has been rarely seen in a recognized pregnancy. There are only three reported cases of partial mosaic trisomy 1 by translocation between the long arm of chromosome 1 and Y, which is similar to our case. Usually, Y-autosome translocation occurs in the Y chromosome breakpoint, which is in the distal euchromatic part (Yq11.23) and in the heterochromatic part (Yq12). Watson et al. (23) reported a monozygotic twin pregnancy at 30 weeks' gestation in which one of the twin was normal and the other had a 46,XY/46,X,-Y,+der(Y)t(Y;1)(q12;q21) with multiple anomalies such as severe hydrocephalus, micrognathia, microstomia, left eye hypoplasia with complete coloboma, hypoplastic cerebellum and macrocephaly. Kaufman et al. (24) described a fetus whose ultrasound at 19 weeks showed an absent calvarium, large omphalocele, possible proboscis, and a suggestion of a VSD and whose karyotype was 46,XY/46,X,-Y,+der(Y)t(Y;1)(q12:q11) (24). Zeng et al. (25) presented a twin following in vitro fertilization at 31 weeks' gestation. The karyotype of twin A was normal, while the others' karyotype was 46,XY/46,X,-Y,+der(Y)t(Y;1)(q12;q12) with left diaphragmatic hernia, hypoplastic lungs, a partial cleft palate, camptodactyly, syndactyly, ventriculomegaly, and hypoplastic cerebellum.
Other chromosomal aberrations associated with CDH include tetrasomy 12p, mosaicism, 9p syndrome, deletion 8, an unbalanced translocation 5q;9p resulting trisomy for 5q and monosomy 9p, supernumber derivatives (22) chromosome abnormality and de Lange syndrome (chromosome 3, q25-2a band region duplication) (26, 27).
Despite the advances in fetal intervention and development of extracorporeal membrane oxygenation, the pregnancy outcome of CDH with chromosomal aberrations has been known as poor. Herein, we present this case of isolated CDH with partial mosaic trisomy 1 diagnosed prenatally and conclude that fetal karyotype must be performed to find any chromosomal aberrations in CDH without additional malformations by prenatal ultrasound, even if the risk for a chromosomal anomaly in isolated CDH seems small.

 XML Download
XML Download