

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ewing's sarcoma (ES) is a small, round, blue cell malignancy that most commonly arises in the skeleton of adolescents and young adults (1). A rare subset of ES, known as extraskeletal Ewing's sarcoma (EES) arises in soft tissue rather than in relationship to bone. EES shares histologic, immunohistochemical, and molecular findings with bone ES. The principal sites of EES are the chest wall, lower extremities, and paravertebral region. The head and neck have seldom been involved as the primary site of EES (2-5). Here we report a case of extraskeletal ES of the hard palate in a young man aged 34 yr. To our knowledge, there is no previously reported instance of this tumor at this site.
CASE REPORT
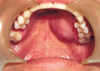
A 34-yr-old male patient was admitted our hospital with a painless swelling of the left hard palate (Fig. 1). It was incidentally detected 4 months previously with a maximum diameter of 3 cm, and did not exhibit ulceration. A paranasal sinus CT-scan revealed a contrast-enhancing mass lesion in left side hard palate with focal bony destruction (Fig. 2).
The patient underwent local excision of the mass lesion. The tissue specimen was fragmented containing several pieces of solid mass and the cut surface was pale tan without showing hemorrhage or necrosis.
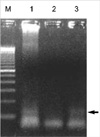
Histological examination of hematoxylin-eosin stained sections showed a homogeneous population of closely packed small neoplastic cells with abundant fibrovascular stroma (Fig. 3). Remnant salivary gland tissue was observed at the tumor's periphery (Fig. 4). Most of the individual cells had scanty cytoplasm, indistinct borders and round or oval nuclei with fine powdery chromatin (Fig. 5). The tumor cells were arranged haphazardly without neural or Homer-Wright rosettes. Mitosis was rare. Some of the tumor cells had a moderate amount of intracytoplasmic glycogen as demonstrated by PAS stain totally abolished after diastase pretreatment. A panel of immunohistochemical stains was performed. The tumor cells were positive for CD99/MIC2 and vimentin and negative for; the leukocyte common antigen, pancytokeratin, cytokeratin 7, neuron-specific enolase, chromogranin, synaptophysin, smooth muscle actin and S-100 protein. The CD99/MIC2 stain exhibited strong membranous staining (Fig. 6). Histologic and immunohistochemical findings were compatible with ES. Ultrastructurally, the tumor cells showed a few intracytoplasmic organelles without evidence of neurosecretory granules or neurofilaments. The RT-PCR showed the presence of the EWS-FLI-1 fusion transcript, which is of the t(11;22) translocation characteristic of ES and primitive neuroectodermal tumor (Fig. 7).
After resection, the patient was treated with multiagent chemotherapy with ifosfamide, cisplatin and etoposide combined with 6,800 cGy of radiotherapy. Eight months later, paranasal sinus CT scan reveals no evidence of recurrence.
DISCUSSION
ES is one of the most controversial tumors. Ever since its first description by James Ewing (6) as a "diffuse endothelioma", remarkable evolution in the concepts regarding its histogenesis and relation with other small round cell tumors, including primitive neuroectodermal tumor (PNET). Recent immunoperoxidase and cytogenic studies indicate that PNET and ES are the same entity showing varying degrees of neuroectodermal differentiation and they are categorized into a group known as the Ewing family of tumors (7, 8).
Some studies require histologic evidence of rosette formation, others require immunohistochemical evidence of neural differentiation, with or without rosettes for a diagnosis of PNET. Schmidt et al. (9) suggested that a tumor was designated a PNET if it had Homer-Wright rosettes on light microscopy or co-expressed two or more neural markers by immunohistochemistry. Using these criteria, they found that patients with PNET had a more aggressive clinical course than those with EES. In our case, tumor cells showed no rosette formation and were negative on immunohistochemical stains for neuron-specific enolase, S-100 protein, chromogranin, and synaptophysin. The tumor cells only showed positive staining for CD99/MIC and vimentin. The CD99/MIC2 identified by the 013 antibody, which is a useful immunohistochemical marker for ES (10). CD99/MIC2 is a cell surface glycoprotein found in virtually all ES and PNET, having been shown in up to 98% of ES and PNET. But it is also detected other small round cell tumors in the differential diagnosis such as T-lymphoblastic lymphoma, poorly differentiated synovial sarcoma, small cell osteosarcoma, rhabdomyosarcoma, desmoplastic small round cell tumor, small cell carcinoma, and Merkel cell carcinoma and it should be used as part of a panel of immunostains, given it's lack of complete specificity (11). The current case could exclude the possibility of other small round cell tumors, such as lymphoma, rhabdomyosarcoma, poorly differentiated synovial sarcoma, by negative immunoreactivities for leukocyte common antigen, desmin, and cytokeratin (9).
Although the exact etiology remains unknown, more than 95% of ES/PNET show the characteristic translocation t(11;22)(q24;q12) or the variant t(21;22)(q22;q12) (7). These translocations fuse the 5'portion of the EWS gene on chromosome 22q12 to either FLI1 on 11q24 or ERG on 21q22. The resultant fusion genes (EWS/FLI1 or EWS/ERG) express chimeric proteins, which are capable of cell transformation and act as aberrant transcription factors. Identification of the fusion transcripts EWS/FLI1 or EWS/ERG by RT-PCR or fluorescent in situ hybridization serves as a sensitive and specific diagnostic test for ES/PNET.
Various therapeutic modalities have been developed (12). Early and confident diagnosis coupled with modern chemoand radiotherapy has greatly improved the prognosis of ES/PNET. This approach has been successful in our patient: surgical excision followed by chemotherapy and radiotherapy resulted in no clinical evidence of recurrence by the nine-month follow-up examination.





 XML Download
XML Download