

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The neuronal migration disorders, X-linked lissencephaly syndrome (XLIS) and subcortical band heterotopia (SBH), also called "double cortex", have been linked to missense, nonsense, aberrant splicing, deletion, and insertion mutations in doublecortin (DCX) in families and sporadic cases (1-8).
Most DCX mutations identified to date are located in two evolutionarily conserved domains (7, 9). The prevalence of DCX mutations is 38-85% in sporadic SBH patients and 100% in all multiplex families with XLIS/SBH (7, 10). Because SBH is typically seen in women with heterozygous DCX mutations and is a less severe phenotype than lissencephaly, which is a typical phenotype of men with hemizygous mutations, SBH is considered a mosaic phenotype that involves lyonization (5). These findings suggest that the conserved domains in DCX play a major role in neuronal migration during neural system development. The precise explanation for the function of the DCX protein in neuronal migration, however, remains unknown.
We performed mutation analysis of DCX in two Korean patients with SBH and report a novel mutation of DCX that is responsible for SBH.
MATERIALS AND METHODS
Patients
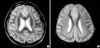
Two unrelated Korean patients with epilepsy and SBH (Patients 1 and 2) and their clinically unaffected family members were studied after obtaining informed consent. The clinical characteristics of the patients are summarized in Table 1. The SBH patients had mild to moderate developmental delays, drug-resistant generalized seizures, and diffuse thin (Fig. 1A) and thick SBH upon brain MRI (Fig. 1B).
Mutation analysis
DNA was extracted from peripheral blood using a standard protocol, and all coding regions of DCX (exons 1 to 7; exon numbering according to RefSeq AJ003112.1, Gen-Bank) were amplified by PCR. PCR assays were performed by using 1.25 U of AmpliTaq polymerase Gold (Applied Biosystem, Foster City, CA, U.S.A.), 100 ng genomic DNA, 2.0 mM MgCl2, and a final concentration of 10 µM for each set of primers (Table 2). Amplification conditions were as follows: an initial denaturing cycle at 95℃ for 7 min followed by 35 cycles of denaturation at 94℃ for 30 sec, annealing at 62℃ for 30 sec, and extension at 72℃ for 1 min. A final extension step of 72℃ for 7 min was used. The PCR products were electrophoresed in a 1.2% agarose gel, and the amplified genomic DNA fragments were extracted and purified using the manufacturer's recommended protocol (QIAquick® gel extraction kit; Qiagen, Germany). Direct sequencing of both strands was performed with BigDye terminator chemistry (PE Biosystems, Foster City, CA, U.S.A.). As the mutation found in Patient 1 destroys an Mnl1 restriction site, restriction fragment length polymorphism (RFLP) analysis was performed using Mnl1 enzyme (New England BioLabs, Beverly, MA, U.S.A.) for the PCR products from both the family members of Patient 1 and 60 unaffected, unrelated Korean women. Samples were then electrophoresed in 4% agarose.
RESULTS
Sequence analysis of the DCX coding region for Patient 1 revealed a c.386C>T change in exon 3 (Fig. 2A). The sequence variation results in a serine to leucine amino acid change at position 129 (S129L), which has not been found in other family members of Patient 1, including maternal grandmother, parents, or siblings. RFLP analysis was conducted to confirm the sequencing result and to exclude the possibility that the genetic variation is a polymorphism. The heterozygous mutant band pattern was observed in Patient 1, whereas it was not found in other family members of Patient 1 or in a control population of 60 healthy, unrelated Korean women (Fig. 2B). No genetic variation in DCX coding region was found for Patient 2 and her mother.
DISCUSSION
In the developing cortex, cortical neurons must migrate long distances to reach the site of their final differentiation. The protein encoded by the DCX gene is a cytoplasmic protein that appears to direct neuronal migration by regulating the organization and stability of microtubules (10). Mutation clusters in the DCX gene were previously identified in exons 2 and 3, and 3 and 4, overlapping significantly with two proposed evolutionarily conserved domains of the DCX protein (7, 9). The first conserved domain binds tubulin and enhances microtubule polymerization, and the second binds tubulin less well and does not enhance microtubule assembly. Both domains seem necessary for optimal DCX protein function, and differences in the location and type of mutations in these regions may produce variable functional disturbances. The mutation identified in the present study is located in the first preserved domain of the DCX protein, which not been reported previously.
In the setting of a family with a single girl affected with SBH, it is hard to determine the risk for SBH or XLIS in future children. Because of X-linkage, a male with a DCX mutation should be clinically affected while a female with the mutation may be unaffected due to germline or somatic mosaicism or skewed X inactivation (5, 11-13). In the present study, the genetic analysis of the DCX mutation c.386C>T was negative in the maternal grandmother, mother, and two siblings of Patient 1 who are free from SBH or XLIS. Thus, it seems most likely that the mutation is sporadic rather than familial. However, predicting the risk to offspring with absolute certainty remains to be clear.
Although Patient 2 in the present study had similar clinical and radiological features to those of Patient 1, she did not carry any genetic variation in the coding region of DCX. This suggests that the non-coding region of DCX, including the promoter region and introns, or other genes implicated in neuronal migration disorders may be responsible for this patient's condition. Further study is required to clarify the pathogenesis of neuronal migration disorders.
We identified a previously unreported missense mutation in the first preserved domain of the DCX protein. This mutation S129L exhibits features suggesting that it is pathogenic. First, like all pathogenic DCX mutations described to date, it is heterozygous. Second, it is absent from a large panel of 120 control X-chromosomes. Third, it occurs in a highly conserved position-the first domain of the DCX protein where the majority of the known DCX mutations cluster. The discovery of the new mutation reported here supports existing evidence that mutation in the first conserved domain of the DCX protein is important in the pathogenesis of SBH.



 XML Download
XML Download