

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Shiga toxin (Stx)-producing Escherichia coli can cause a range of illnesses from self-limiting watery diarrhea and hemorrhagic colitis to severe conditions such as hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (1). Stx binds to cell-surface glycosphingolipids, terminating in galactose-α1,4-galactose, whereupon the complex is internalized, with resultant inhibition of peptide elongation (2, 3). The major glycosphingolipid that binds Stx is galactose-α1,4-galactose-β1,4-glucose ceramide (globotriaosylceramide: Gb3) in the case of Stx-1 and Stx-2, and globotetraosylceramide (Gb4) for Stx-2e. These toxins are structurally similar heterodimers composed of one enzymatically active A subunit (4) that irreversibly inhibits protein synthesis by means of its N-glycosidase activity on ribosomal subunit and B subunit pentamer that binds Gb3 (5, 6).
Epigallocatechin-3-gallate (EGCG), a major ingredient of green tea, has been known to have a variety of physiological functions, such as anti-carcinogenic, anti-oxidant, anti-angiogenic, anti-viral (7-9), and anti-bacterial activities (10-12). Recently, others and we have showed that EGCG suppresses mitogen-activated protein kinase (MAPKs), and NF-κB activation (12, 13), and lipooxygenase and cyclooxygenase (COX) activities (14), and arrest the cell cycle (15) in tumor cells.
Differentiated intestinal epithelial cells express Gb3 (16), and are sensitive to toxin-mediated cytotoxicity. Stx produced in the intestine can be translocated to the bloodstream without cellular damage and can reach renal endothelial cells causing HUS (17, 18). Several studies support the idea that Stx is important in the pathogenesis of bloody diarrhea that is caused by damage of the intestinal epithelium. And recent study has also demonstrated that invasion of the intestine in vivo or of intestinal cells in vitro by Stx produces augmented release of intestinal cytokines including IL-1, TNF-α, IL-4, and IL-10, suggesting that these cytokines may be important in the pathogenesis of inflammatory diarrheal disease (19-22). Especially, TNF-α and IL-1 have been known to be important cytokines to induce Gb3 and we here confirmed that TNF-α increases Gb3 synthesis through the upregulation of ceramide glucosyltransferase (CGT), lactosylceramide synthase (GalT2), and Gb3 synthase (GalT6) (23-26).
In this study, we investigated whether EGCG can suppress TNF-α induced Gb3 production, and EGCG has the potential to modulate Gb3 production through MAPKs (p38, c-Jun N-terminal kinase (JNK)1/2, and ERK1/2), NF-κB activation, and transcription of Gb3 synthesis enzymes in human colon epithelial HT29 cells. To accomplish this, we exposed intestinal cell lines to EGCG in vitro and analyzed the expression of Gb3. We found that EGCG inhibits TNF-α induced Gb3 synthesis by interfering with the MAPKs and NF-κB pathways in HT29 cells.
MATERIALS AND METHODS
Cell culture
A HT29 cell line was obtained from Korean Collection of Cell Cultures (Seoul, Korea). HT29 cells were grown in RPMI Medium 1640 (Gibco BRL, Gaithersburg, MD, U.S.A.) supplemented with-glutamine, 25 mmol/L of HEPES buffer, and sodium bicarbonate containing 10% fetal bovine serum (FBS), penicillin, streptomycin sulfate, and sodium hydrogen carbonate. The cells were incubated in humidified 5% CO2 atmosphere at 37℃. EGCG was dissolved in dimethylsulfoxide (DMSO). DMSO (0.01%), used as vehicle alone, did not affect HT29 cells. Recombinant human TNF-α (R&D Systems Minneapolis, MN, U.S.A.) was used as a stimulator of HT29 cells. For all the studies carried out, cells were seeded at a low density (5×106 cells/mL) in 12 well or 100 mm dishes. The cells were allowed to attach overnight and rendered quiescent through removal of feeding medium and its subsequent replacement with a serum-free medium 24 hr before the addition of experimental media. And after the treatment of EGCG, the media was changed daily to maintain the EGCG effect.
Apoptosis assay
Annexin V/PI staining was used to evaluate the survival of the HT29 cells. Following specified treatments, the cells were collected, washed with phosphate-buffered saline (PBS) and resuspended in 100 µL of annexin binding buffer (140 mM NaCl, 10 mM HEPES, pH 7.4, 25 mM CaCl2). Then, 5 µL of annexin V-FITC conjugate (Biosource International, Ridlington, Oxford, U.K.) and 2 µL of propidium iodide (1 mg/mL) were added, and this suspension was incubated for 15 min at room temperature. The samples were then further diluted with 400 µL of annexin binding buffer. The cells were analyzed by FACSCalibur (BD Biosciences, San Jose, CA, U.S.A.). All of the annexin V-positive cells were considered apoptotic (early and late), and their percentage among the total number of cells was calculated. PI+ Annexin V- cells were considered necrotic.
Flow cytometric analysis
The pure Stx-IB subunit was dialyzed in a 0.1 M sodium carbonate buffer (pH 7.4) for 24 hr. Fluorescein isothiocyanate (FITC; Molecular Probes, Leiden, The Netherlands) was added to 50 µg/mg of the B subunit. The complexes were incubated at room temperature for 1 hr. The unreacted FITC was removed by a process of gel filtration (Sephadex G25). After 24 hr, the culture in RPMI 1640 containing 0.5% FBS, HT29 cells were incubated with 0.5 µL of Stx-IB-FITC on ice for 30 min. The stained cells were analyzed using a FACSCalibur.
RNA extraction and reverse transcription-PCR
Total RNA was isolated from the harvested cells using the TRIzol reagent (Gibco BRL). The concentration of total RNA in the final elute could determine by spectrophotometry. The total RNA (2 µg) was converted to cDNA by reverse transcriptase at 37℃ for 90 min using first-strand cDNA synthesis kit (Amersham Pharmacia Biotech, NJ, U.S.A.). The PCR profile included a 94℃ denaturalization for 5 min followed by 27 cycles of denaturalization at 94℃ for 30 sec, annealing at 50℃ for 30 sec, extension at 72℃ for 30 sec, and a final extension at 72℃ for 10 min. The primers used in this study were as follows: for β-actin (GenBank accession no. NM001101), forward primer 5'-CAAGAGATGGCCACGGCTGC-3'and reverse primer 5'-GCCAGTAGAGGCAGGGATGATGTTC-3', 251 bps, which yields a product size of 274 bps; for ceramide glucosyltransferase (GenBank accession no. D50840), forward primer 5'-CTTCTGAAACCACTGAAAGG-3'and reverse primer 5'-CAACTTCATATCCTGGCATT-3', 240 bps; for GalT6 (GenBank accession no. AB037883), forward primer 5'-GGATGCTTTGTTCTTTTCTG-3'and reverse primer 5'-TTAAAATGCTTCTCCTGAGC-3', 239 bps; for GalT2 (GenBank accession no. AF097159), forward primer 5'-TCAACGGTACAGATTATCCC-3'and reverse primer 5'-TTCCATCTGGGTTTACAGTC-3', 249 bps; for α-galactosidase (GenBank accession no. NM_000169), forward primer 5'-CTTCTGAAACCACTGAAAGG-3'and reverse primer 5'-CAACTTCATATCCTGGCATT-3', 253 bps. The final PCR products were loaded on 1.5% agarose gels.
Western blot analysis for MAPKs
Twenty-five µM EGCG-pretreated HT29 cells (5×106/mL) were stimulated with TNF-α (50 ng/mL). The cells were rinsed twice with ice-cold PBS and harvested by scraping in 1 mL of ice-cold PBS. The pellets were resuspended in 100 µL of lysis buffer (iNtRON Biotech, Korea) and incubated on ice for 30 min. The lysates were removed by centrifugation at 15,000×g for 10 min. The samples were immediately aliquoted and stored frozen at -70℃. Protein concentrations were determined using a bicinchoninic acid protein assay (Sigma, St. Louis. MO, U.S.A.). Thirty µg of protein (homogenate) was separated by 10% SDS-polyacrylamide gels and transferred to PVDF membranes (Millipore, MA, U.S.A.). After blocking with 5% skim milk, the membranes were incubated with antihuman-phospho-ERK antibody, antihuman-phospho-JNK antibody, and antihuman-phospho-p38 antibody (Santa Cruz Biotech, CA, U.S.A.) for 12 hr at 4℃ and then with anti-IgG-horseradish peroxidase (DAKO, High Wycomde, Bucks, U.K.) for 1 hr. Epitopes on proteins recognized specifically by antibodies were visualized by using enhanced chemiluminescence (ECL) detection reagents (Amersham Pharmacia Biotech). After stripping, the membranes were reprobed with antihuman ERK antibody, antihuman JNK antibody, and antihuman p38 antibody (Santa Cruz Biotech) as the respective loading controls.
Extraction of cytosolic and nuclear proteins and Western blot analysis for NF-κB
Cytosolic and nuclear extracts were separated by 10% SDS-PAGE to detect NF-κB, respectively, and transferred to PVDF membranes (Millipore). After blocking with 5% skim milk, NF-κB membranes, respectively, were incubated with antihuman NF-κB (p65) (Santa Cruz Biotech) for 12 hr at 4℃. Membranes were incubated with the anti-IgG-horseradish peroxidase (DAKO) for 1 hr. Epitopes recognized specifically by antibodies were visualized by using ECL detection reagents.
Statistical analysis
The results were expressed as the mean±S.E. for the number of experiments. The statistical significance was compared among each treated group and the control by a Student's t-test. Each experiment was repeated at least three times and yielded comparable results. Values with p<0.05 were considered significant.
RESULTS
EGCG was non-toxic and non-apoptotic to HT29 cells in vitro
EGCG has been reported to reduce cell proliferation and to induce cell apoptosis in some tumor cell lines (27), with the TNF-α pathway is also linked to apoptosis via JNK/P38. We experimented the effect of EGCG and TNF-α to prove whether Gb3 inhibition by EGCG is associated with cell apoptosis in cultured HT29 cells. Subconfluent dishes of HT29 cells were treated with EGCG (25 µM), TNF-α (50 ng/mL) and EGCG with TNF-α for 48 hr, and apoptosis was measured by FACS. Those concentrations did not affect cell viability and did not induce apoptosis (Fig. 1).
TNF-α stimulated the expression of Gb3 in HT29 cells
Dose-response analyses for TNF-α from 10 to 100 ng/mL and for the time course from 12 hr to 72 hr were performed. TNF-α increased the Gb3 expression in HT29 cells in a time- and concentration-dependent manner (Fig. 2).
EGCG suppressed Gb3 expression in HT29 cells
Then, it was investigated whether or not EGCG would affect the TNF-α induced Gb3 expression in HT29 cells. The HT29 cells were pretreated with four doses of EGCG (10, 25, and 50 µM) for 2 hr and then stimulated with TNF-α for 2 days, and Gb3 expression was examined by FACS. Gb3 induction was derived by TNF-α (50 ng/mL) and significantly decreased in cells treated with EGCG 25 and 50 µM (Fig. 3B). DMSO (0.01%) used as vehicle did not affect on the Gb3 induction from TNF-α-stimulated HT29 cells (data not shown). TNF-α increased the Gb3 content on the HT29 cells about 2.5 folds, and remarkably decreased the Gb3 content in cells pretreated with 25 and 50 µM of EGCG.
EGCG inhibited TNF-α-induced MAPK phosphorylation
To determine if EGCG has the ability to block the activation of MAPKs, the HT29 cells were pretreated with EGCG 2 hr prior to TNF-α stimulation. Our results showed that EGCG inhibits all three MAPK subgroup, ERK, JNK1/2, and p38-MAPK, in TNF-α-pretreated HT29 cells, without affecting the total levels of these kinases (Fig. 4). A scanning analysis of the blots revealed that the phosphorylation inhibitory effect of EGCG was more evident. These results indicate that MAPK phosphorylation was inhibited by EGCG pretreatment.
Effect of EGCG on TNF-α-induced NF-κB nuclear translocation
It has been shown that TNF-α activates a variety of signal pathways, including that of NF-κB (24), which plays a critical role in regulating various genes. To determine whether EGCG blocks TNF-α-induced activation of NF-κB, a nuclear extract was prepared from HT29 cells treated with TNF-α and EGCG; nuclear translocation of the NF-κB, p65 subunit was detected by western blot. The TNF-α-mediated NF-κB (p65) increase in the nuclei was blocked by the treatment of EGCG (Fig. 5A).
EGCG suppressed TNF-α-induced CGT, GalT2, and GalT6 mRNA expression
Gb3 synthesis is associated with three glycosphingolipid enzymes (Fig. 6). Each of the three enzymes involved in Gb3 synthesis (CGT, GalT2, and GalT6), as well as the enzyme involved in Gb3 degradation (α-galactosidase), was evaluated. This set of experiments investigated the mechanisms responsible for the inhibition of TNF-α-stimulated Gb3 expression in HT29 cells. CGT, GalT2, and GalT6 mRNA in-EGCG (25 µM) pretreated HT29 cells were analyzed by RT-PCR. CGT, GalT2 and GalT mRNA accumulation was induced by TNF-α and remarkably decreased in cells pretreated with EGCG (25 µM) (Fig. 7). These finding indicate that EGCG (25 µM) suppresses CGT, GalT2, and GalT6 mRNA levels that are related to Gb3 synthesis. In contrast, α-galactosidase activity was not changed by TNF-α alone, and was co-treated with EGCG.
DISCUSSION
Infections with Stx-producing E. coli have been known to induce intestinal cytokines including IL-1 and TNF-α. These cytokines play an important role in the interactions of bacteria with mucosal surface and are known to increase the binding of E. coli to intestines (28) and the Gb3 content in intestinal cells. And during the development of HUS, Stx may work cooperate with LPS and the LPS-induced cytokines TNF-α and IL-1 to damage vascular endothelial cells and intestinal epithelial cells.
It has been demonstrated that TNF-α, IL-1β, and bacterial LPS require 24 to 48 hr to maximally sensitize endothelial cells to the Stx, but intestinal glycoconjugates have not been clearly demonstrated to be involved in Gb3 induction by these cytokines or LPS (25, 26).
Accordingly, our first set of experiments was designed to confirm that the inflammatory cytokine TNF-α can induce Gb3 production. We have demonstrated here that the incubation of HT29 cells with TNF-α markedly enhances the Gb3 contents in the cells.
The effects of EGCG on the activation of MAPK members in selected cell lines have also been reported. It was shown that treatment with EGCG inhibited the ultraviolet-B-induced activation of p38-MAPK in the human keratinocyte cell line (29), whereas other studies showed have shown that EGCG activated all three MAPK families including the extracellular signal-regulated kinase (ERK1/2), JNK, and p38 in HeLa cells (30). In vascular smooth muscle cells, EGCG inhibited the platelet-derived growth factor-β-induced activation of ERK1/2 in a dose-dependent manner (15). In addition, EGCG selectively inhibited the interleukin-1β-induced activation of JNK, but not ERK1/2 or p38 MAPK, in human osteoarthritis chondrocytes (31). Thus, it appears that the MAPK-activating or inhibitory effects of EGCG may be stimulus- and/or cell type-dependent (13).
EGCG has been studied to make use of its biological effects including anti-oxidant, anti-proliferative, anti-cancer (7-9), and anti-bacterial activities (10-12). Interestingly, EGCG has the potency to inhibit production and/or release of Stx from EHEC cells (32). In this study, we examined the effect of EGCG on Gb3 production and its mechanism of action in regulating the MAPKs and NF-κB pathways in HT29 cells. We observed that the pretreatment of EGCG resulted in the inhibition of TNF-α-mediated MAPK phosphorylation and NF-κB activation.
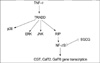
Also, TNF-α has been known to increase the transcription of CGT and GalT2,6 enzymes (23) that are related to the Gb3 production pathway in eukaryotic cells. The promoters of these three enzymes contain an NF-κB-recognition site (25). EGCG also decreased the enzymes related to the Gb3 synthesis pathway via the transcription level regulation of NF-κB activation. Based on our results, we proposed a hypothetical scheme explaining EGCG actions on Gb3 production in Fig. 8.
Taken together, our data clearly indicates that EGCG inhibits TNF-α-mediated Gb3 synthesis by blocking both the MAPKs and the NF-κB pathways in HT29 cells. Thus, it is plausible to propose that EGCG regulates the Gb3 expression of host colonic cells in situ and that consumption of EGCG may alter the interaction of Stx family toxins with the mucosa and reduce disease expression in humans, especially during seasonal food-poisoning periods.







 XML Download
XML Download