

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Amyloidosis is a connective tissue disease characterized by the deposition of insoluble, fibrous amyloid proteins, primarily in the extracellular space of organ tissues (1). Although median nerve compression, or carpal tunnel syndrome, is a frequent finding of AL amyloidosis, femoral nerve compression caused by hypertrophic amyloid myopathy has not been previously described (2).
We report a case of a patient with AL amyloidosis who developed bilateral lower extremity weakness in association with femoral nerve compression due to iliopsoas pseudohypertrophy.
CASE REPORT
A 54-yr-old man visited our clinic because of lower extremity weakness and polyarthritis involving the shoulders, wrists, and hands. He had been in good health until 2 months prior to visiting our clinic, when he began to experience anorexia and fatigue. At that time, he was diagnosed at a local hospital as having end stage renal disease (ESRD) of unknown etiology and was started on regular hemodialysis. About a week after initiating hemodialysis, he had pain and swelling in both shoulders, wrists and metacarpophalangeal (MCP) joints, followed by progressive bilateral lower extremity weakness, which caused him to visit our clinic. He also began to have a tingling sensation in both hands.
On admission, his blood pressure was 135/88 mmHg, heart rate 94/min, respiration rate 20/min, and body temperature 36.2℃. A physical examination showed severe swelling in both hands and feet, and tenderness at wrists, MCP joints, and shoulders. Bilateral shoulder pads were also present. Severe, isolated bilateral knee extensor weakness (muscle strength grade I) was detected, with relatively preserved strength of knee flexors and of hip and ankle flexors and extensors (grade IV to V). A decreased sensation in medial thighs was noted. Diffuse, firm swelling of bilateral inguinal area was detected. Deep tendon reflex in both knees was reduced, as was grasping power in both hands. Laboratory data showed anemia (hemoglobin of 8.3 g/dL), azotemia (creatinine of 6.7 mg/dL), and elevated ionized calcium level of 1.55 mEq (normal 1.05-1.35). The serum total protein and albumin levels were 5.7 g/dL (normal 6.0-8.0) and 3.4 g/dL (normal 3.3-5.2), respectively. The serum creatine kinase level was 117 IU/L (normal 20-270), lactate dehydrogenase, 287 IU/L (normal 100-225), and beta 2-microglobulin, 52.9 mg/L (normal 1-3). Erythrocyte sedimentation rate of 76 mm/hr (normal 0-10) and C-reactive protein level of 2.0 mg/dL (normal 0-0.5) were measured. Urinalysis with microscopy showed three positive amount of albumin excretion and normal sediment findings. Serum anti-nuclear antibody and rheumatoid factor were negative. An electrocardiogram showed normal sinus rhythm without any particular abnormalities. Simple radiographic films of the shoulders, hands, feet and the whole spine failed to show any specific bony abnormalities, but did show diffuse systemic osteopenia and soft tissue swelling in both hands and feet. Ultrasonography revealed bilaterally contracted kidneys.
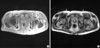
Amyloidosis was confirmed by the shoulder pad biopsy, which showed an apple green birefringence under polarized light when stained with Congo red and the characteristic non-branching fibrillar structure of amyloid by electron microscopy. Immunofluorescent staining (IF) for kappa light chain was strongly positive (Fig. 1). Negative M spike in serum/urine protein electrophoresis, abnormal kappa chain arc in serum/urine immunoelectrophoresis, and bone marrow plasmacytosis (12.9%) suggested kappa-AL amyloidosis associated with plasma cell dyscrasia. There was no evidence of muscular amyloid deposition in the quadriceps biopsy, ruling out amyloid deposition in quadriceps muscle as the cause of knee extensor weakness. Subsequent bilateral femoral nerve conduction study (NCS) and vastus lateralis/medialis electromyography showed typical neuropathic findings (decreased compound muscle action potential (CMAP) amplitude, abnormal spontaneous activity, discrete interference pattern). Median NCS revealed no formation of CMAPs and sensory nerve action potentials, in contrast with intact ulnar NCS. These findings indicated bilateral femoral and median nerve palsies as the causes of the weakness in knee extensors and of the grasping difficulty in hands. To identify the culprit lesions causing nerve palsies and to evaluate the painful shoulder joints, we obtained magnetic resonance images (MRI) of the wrists, pelvis and shoulders. The MRI findings showed bilateral wrist joint fluid accumulation, median nerve compaction in a swollen carpal tunnel, intrinsic hand muscle swelling, marked iliopsoas hypertrophy, and prominent subdeltoid bursa thickening. Iliopsoas muscle showed heterogeneous intensity in both T1 and T2 weighted images with irregular gadolinium enhancement in T1 weighted images (Fig. 2). The neurovascular bundle containing the femoral nerve was medially adjacent to the iliopsoas muscle, which seemed to compress the bundle. We were unable to find subcutaneous fat reticulation at the mid-thigh level, previously reported in the few cases of MRI findings in amyloid myopathy.
A month after admission, we performed bilateral carpal tunnel release and psoas muscle excision, to decompress the nerves. During the operation, tight adhesion was found between the femoral nerves and the psoas muscles. All surgical specimens gained from carpal tunnel release and psoas muscle excision showed an apple green color under polarized light with Congo red staining and showed the same characteristic fibrillar structure of amyloid by electron microscopy, as was found in the shoulder pad biopsy. In muscle specimens, deposits were primarily distributed in the perimysium, endomysium and perivascular space with scanty inflammatory cells (Fig. 3). Variable-sized muscle fibers were frequently separated by a thickened interstitium. About three weeks after the operation, he could grasp with both hands and his knee extensor strength improved gradually to grade III. We planned to try chemotherapy consisting of dexamethasone and cyclophosphamide after the wound healed.
A month after the operation, he began to suffer profuse diarrhea due to intestinal amyloidosis. His diarrhea was refractory despite one cycle of chemotherapy. Subsequently, serious malnutrition ensued. He died of severe cachexia and combined infection six weeks after the chemotherapy.
DISCUSSION
The involvement of skeletal muscles by AL amyloidosis can be divided into two phenotypes. One is characterized by proximal muscle atrophy and presents with inflammatory myositis- like features (3, 4). The other is characterized by muscle pseudohypertrophy, macroglossia, or palpable intramuscular masses without inflammatory infiltration or elevated muscle enzymes (5, 6). The present case belongs more to the latter form. However, it is distinctive in that muscle weakness was associated with compressive neuropathy rather than amyloid myopathy itself.
Amyloid arthropathy in multiple myeloma is known to be rare and presents in the form of shoulder hypertrophic arthropathy or of rheumatoid arthritis-like peripheral arthritis (7). The polyarthritis in this case, resulting from amyloid deposition in the periarticular tissues, showed both forms of arthropathy by involving both shoulders, wrists, and MCP joints. Although the marked periarticular amyloid deposition seen in this case, together with his ESRD status, reminded us of dialysis associated amyloidosis, this seemed to be unlikely because the duration of dialysis (< 2 months) was too short for this type of amyloidosis and because clinically relevant visceral involvement, which manifested as intestinal amyloidosis in this patient, is known to be rare in dialysis associated amyloidosis, and to be largely confined to long term patients with more than 15 yr of dialysis (8). The positive IF for kappa light chain in the shoulder pad biopsy confirmed this speculation.
Carpal tunnel syndrome seen in this case, is not only a frequent finding of AL amyloidosis, but also one of the most common features of hereditary amyloidosis which is characterized by a family history (autosomal dominant), and in most cases, sensorimotor polyneuropathy (2). The diagnosis of hereditary amyloidosis is often missed even though different forms of hereditary systemic amyloidoses has been found in as many as 9.7% of patients who were initially classified as having AL amyloidosis and though the prognosis can be improved with liver transplantation (9, 10). A Korean case of hereditary amyloidosis, as a form of familial amyloid polyneuropathy, has been reported (11). In our case, however, the definite documentation of plasma cell dyscrasia, no clinical penetrance of polyneuropathy in a family history as well as in his manifestations, and finally, the positive IF demonstrating kappa light chains as a precursor protein made it possible to exclude hereditary amyloidosis as the underlying process of the disease.
In this case, light chain deposition disease might have been co-present with AL amyloidosis, and played a role in renal failure, because strong and diffuse immunofluorescence, which is unusual in AL amyloiosis, is a finding consistent with light chain deposition disease, as is kappa rather than lamda light chain involvement (12).
In summary, we present a rare case of AL amyloidosis with femoral nerve compressive neuropathy, hypertrophic amyloid myopathy and amyloid arthropathy. It should be emphasized that various musculoskeletal involvements including polyarthritis and proximal muscle weakness constitute elusive manifestations of amyloidosis and that not only amyloid myopathy but also compressive neuropathy should be included in the differential diagnosis of muscle weakness in amyloidosis, especially when muscular hypertrophy is present.


 XML Download
XML Download