

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Fulminant hepatic failure carries a very high mortality of 80-90% (1). Orthotopic liver transplantation has now become the treatment of choice of fulminant hepatic failure, with a survival rate of 50-75% after 1 yr (2). But, spontaneous survival rate of patients with fulminant hepatic failure ranges from 15% to 25% (3). Therefore, a liver assist device to help the liver recover spontaneously can be an alternative method to replace orthotopic liver transplantation or a bridge to patients waiting for a suitable donor in preventing the occurrence of irreversible neurologic damage. Several devices have been developed and employed clinically.
prove the functional support of liver assist devices in fulminant hepatic failure, it is necessary to establish a standardized hepatic failure model which closely correlates with clinical situations and can be reproduced in large animals. There are several animal models of hepatic failure. The surgical models include total hepatectomy, partial or complete devascularization, and portacaval shunt with 70% partial hepatectomy (4-7). The pharmacological models include D-galactosamine, acetaminophen, and carbon tetrachloride (4, 8-12). The aim of these models is to simulate acute fulminant hepatic failure that results in the rapid onset of hepatic coma and death. Even though the application of liver assist devices in human has been performed, the additional development of technology and demonstration of safety, efficacy, and mechanism of action should be greatly accelerated by testing a clinically relevant large animal model.
Ischemic injury is one of the reasons for hepatic failure (6). For pigs, the tolerable period of hepatic ischemia varies from 35 to 180 min (6, 14-16). If the time of hepatic ischemia exceeds the tolerable period, hepatocytes can be injured irreversibly and necrosis can occur, too. In the present study, we have evaluated a model of fulminant hepatic failure induced by clamping the portal vein and the hepatic artery intermittently to cause reperfusion injury to the liver and then by ligating hepatic artery at the end of the last clamping.
MATERIALS AND METHODS
Animals
Female white pigs, weighing from 17 kg to 25 kg, were used for this experiment. One day before surgery, 20 g magnesium sulfate was administered orally for bowel cleansing and then pigs were fasted. All the experiments were conducted under the supervision of a veterinarian according to local institutional guidelines for the care and use of laboratory animals.
Operative procedure
All operative procedures were performed at the "Clinical Research Institute, Seoul National University Hospital. Premedication was performed with an intramuscular injection of ketamine chloride (25 mg/kg). After injecting Pentothal sodium (2 mg/kg), the animals were intubated and ventilated with a mixture of nitrous oxide, oxygen, and enflurane. One catheter was placed in the external jugular vein for infusion and another catheter was introduced into the femoral artery for both blood pressure monitoring and blood sampling. The abdomen was opened by a midline incision. The common bile duct, the hepatic artery, and the portal vein were exposed. A sling was laid around the hepatic artery and the portal vein for clamping. It took 25 min for the 1st and 2nd clamping and 30 min for the 3rd and 4th. The hepatic artery was ligated after the 4th clamping (ligation group, n=5). Pigs whose hepatic artery was not ligated after clamping were assigned to the non-ligation group (n=5). The intervals between each clamping were 30 min and total clamping time was 110 min.
During the operation, lactated Ringer's solution was given and 8.4% NaHCO3 and calcium gluconate were administered for the maintenance of physiologic status. Five furosemide injections (10 mg) were totally given every 90 min to make them excrete potassium. The anesthesia was discontinued after the operation was over. The animals were kept in isolated cages and carefully controlled and their behaviors were registered.
Clinical assessment
The behaviors of the animals after awaking from anesthesia were controlled continuously. The following criteria were registered: abilities to get up, to walk, and to respond to painful stimuli, and also light reflex of pupils. The purpose of these observations was to detect the signs of hepatic encephalopathy.
Biochemical measurements
Blood sampling was done four times: before clamping, after 4 hr and 7 hr since the first clamping respectively, and lastly before death (in non-ligation group, 20 hr after clamping). And the following parameters were measured from the experiment: glucose, aspartate transaminase (AST), alanine transaminase (ALT), total bilirubin, blood urea nitrogen (BUN), creatinine, ammonia, potassium, lactic acid, prothrombin time (PT) and pH. The PT was not checked in the time when it has passed 7 hr since the first clamping.
Histologic evaluations
Specimens of the liver were taken three times: before clamping, after 4 hr since the first clamping, and immediately after death (in non-ligation group, 20 hr after clamping). By means of liver biopsy, liver tissues were fixed by 1 cm thickness in 10% formalin. After fixation, 27 µm sections of each biopsy were stained with hematoxylin eosin and examined by light microscopy
RESULTS
Survival
In the ligation group, the first animal died from hyperkalemia at 6.5 hr after ligation of the hepatic artery. The reason was that 8.4% NaHCO3 and calcium gluconate were not administered. The remaining 4 animals were administered and died after the duration of 14.4±2.1 hr (range: 12.5-17.0). In the non-ligation group, all five animals have survived for more than 7 days.
Clinical assessment
The animals in the non-ligation group did not show marked behavioral abnormalities. The first abnormality observed in the ligation group was usually ataxic gait and impaired balance. And then drowsiness, a decrease in pain sensations, and the posture like walking in the air spontaneously at supine position were followed in 10 hr after ligating the hepatic artery. The loss of sensation, absence of light reflex, and nonreactive coma developed after 10 hr since ligating the hepatic artery.
Biochemical assessment
Metabolic acidosis was observed after intermittent clamping of the portal vein and the hepatic artery, but it was corrected by administration of sodium bicarbonate. In the non-ligation group, the levels of ammonia, lactic acid, AST, ALT, and creatinine gradually declined after showing increased values initially, whereas BUN and potassium remained within the normal range. In the ligation group, the levels of ammonia, lactic acid, and creatinine showed a progressively increasing pattern. The levels of ammonia, lactic acid, total bilirubin, and BUN increased after 4 hr of hepatic ligation. However, the difference were not significant statistically. The increase of AST, ALT, creatinine, and potassium was not significant statistically, either. PT recovered normal range after slight prolongation in the non-ligation group, but it was gradually prolonged in the ligation group (Fig. 1, 2, 3, 4).
Histopathology
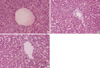
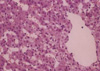
The histologic features in the non-ligation group which were checked before clamping, after 4 hr and 20 hr since the first clamping, respectively were shown in Fig. 5. There were no histologic differences: widened portal spaces with infiltration of a few inflammatory cells, ischemic changes of the hepatocytes in porto-to-portal space, and dilated sinusoidal spaces. The findings obtained after 4 hr since ligating the hepatic artery (Fig. 6) were similar to those of the non-ligation group but nuclear pyknosis and cytoplasmic condensation were found in focal area. At autopsy, the histologic features mentioned above were also shown and furthermore parenchymal hemorrhage, necrosis, nuclear pyknosis and cytoplasmic condensation in a multifocal area were shown (Fig. 7).
DISCUSSION
In the orthotopic liver transplantation, transient ischemia and reperfusion injury is one of the reasons of graft failure (13). Nitta et al. (16) reported that impairment of hepatic energy metabolism by intermittent portal triad cross-clamping is mainly due to reinflow of pooled-portal blood to the previously ischemic liver rather than hepatic warm ischemia. Li et al. (17) demonstrated that in the simple model of orthotopic hepatic autotransplantation in case of dogs, the extension of hepatic arterial ischemic time showed obvious ultra-microstructural changes in the liver and biliary duct, and the levels of serum bilirubin and ALT were elevated. The ischemia and reperfusion injury was represented in this model and splanchnic venous congestion was resolved by declamping the portal triad. Although several investigators verified that ischemia and reperfusion injury was the main cause of hepatic failure in the experimental animal studies, additional hepatic artery ligation was needed to develop hepatic failure in our model.
Terblanche et al. (4) suggested that five criteria should be met for a good animal model of hepatic failure: 1) reproducibility, 2) potential reversibility, 3) the agent used to produce liver failure should cause selective liver damage, 4) use of a large animal, and 5) the lack of relevant biohazard. In the present study, the surgical experimental model of liver failure was reproducible. It has supported original anatomy of the portal vein and bile duct systems. Therefore liver regeneration can be possible if a liver assist device is applied. Hepatic coma was induced by massive hepatocytes necrosis and death could be attributed to progressive liver failure. The mortality rate was 100% when the hepatic artery was ligated after the intermittent clamping and the declamping of the portal triad. The period before death was long enough to allow the use of liver assist devices. Therefore, all requirements for ideal hepatic failure model except reversibility are fulfilled in this study.
In our model, the plasma level of ammonia increased, although it was not significant statistically. Ammonia is a neurotoxin which may precipitate hepatic encephalopathy. In the brain, ammonia may induce changes of blood-to-brain transportation of amino acids and alterations of brain energy metabolism. Furthermore, ammonia directly alters neuronal electric activity by inhibiting the generation of both excitatory and inhibitory post-synaptic potentials (18). Hyperammonemia is typical of fulminant hepatic failure and it indicates an impaired detoxification system.
In the ligation group, the levels of total bilirubin, AST and ALT increased progressively. Lactic acid increased abruptly and then progressively. It suggested a failure of hepatic clearance of peripherally generated lactic acid in the shock state (8). PT was gradually prolonged in the ligation group. Coagulopathy occurred as a result of decreased synthesis of clotting factors and disseminated intravascular coagulation (19). The transient increase in the level of blood glucose after clamping the portal vein and the hepatic artery in two groups was considered as a stress reaction. And it was believed for the increase to be caused by the mobilization of glycogen stores. Hypoglycemia which is a characteristic of fulminant hepatic failure was seen in the ligation group. It has been shown from the result of inhibiting both gluconeogenesis and glucose release from the liver into the blood stream and failure of insulin metabolism (11). But hypoglycemia is not a major factor to bring on coma or death.
This model also showed features of hepatic encephalopathy by inducing massive necrosis of hepatocytes and releasing the toxic by-products into systemic circulation through the portal vessel and bile duct systems. In the ligation group, except for the first animal, the other four pigs showed the posture like spontaneously walking in the air at supine position. The animals lost pain sensation for some hours before death and even pupils did not respond to light for some hours before death. This neurological changes are similar to those of fulminant hepatic failure model in pigs induced by D-galactosamine or graded devascularization (5, 9).
In conclusion, our new model is similar to the hepatic failure in human beings according to the clinical, laboratory and pathologic findings. It may be the advantage of this model that we can make it technically easily, which makes it highly reproducible. To develop fulminant hepatic failure, it is necessary not only to induce the ischemia and reperfusion injury to the liver parenchyma by intermittent clamping and declamping the portal triad, but also to ligate the hepatic artery. This model can be used to test the efficacy of liver assist devices.
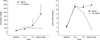




 XML Download
XML Download