

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Acute lymphoblastic leukemia (ALL) is the most common malignancy in childhood, accounting for approximately 25% of all pediatric malignancies. More than 250 children are newly diagnosed ALL every year in Korea. It is well established that the identification of cytogenetic abnormalities is very useful for the prediction of outcome in childhood ALL. For example, t(9;22), 11q23 abnormalities, and hypodiploidy are known to confer a poor prognosis, and t(12;21), hyperdiploidy are associated with a favorable outcome (1). The association between the deletion of 9p21 and poor prognosis was suggested (2-4). However, questions about the relationship between t(12;21) and good prognosis have arisen because several patients with TEL/AML1 fusion showed poorer clinical outcome if other gene rearrangements coexist (5, 6).
Although detection of chromosome aberrations in ALL has been improved by the development of cytogenetic techniques in conventional G-banding analysis, prognostically important structural or numerical chromosome aberrations may frequently go undetected using conventional G-banding alone due to poor chromosome morphology and few malignant metaphases (7, 8). Especially, 9p abnormalities, t(12;21), and some of the 11q23 rearrangements are very difficult, or even impossible, to detect by conventional G-banding analysis. Interphase fluorescence in situ hybridization (FISH) is a rapid and sensitive tool for detecting gene rearrangements, and thus has been frequently used at the time of diagnosis and during follow-up of patients to monitor minimal residual disease.
There have been few Korean studies evaluating the incidences of BCR/ABL, MLL, TEL/AML1, and p16 gene rearrangements commonly found in childhood ALL because FISH has not yet been routinely used at diagnosis of childhood ALL. Therefore, we cannot confirm the results suggesting the existence of geographical variations in the incidence of TEL/AML1 fusion (9, 10).
In the present study, we performed FISH with probes for BCR/ABL, MLL, TEL/AML1 rearrangements, and p16 deletions for each case of childhood ALL. The aims of this study were to estimate the incidences of different genetic subgroups with abnormalities involving the above genes in Korean childhood ALL, to identify new abnormalities, and to demonstrate the usefulness of FISH.
MATERIALS AND METHODS
Patients
Among patients diagnosed childhood ALL between 1997 and 2002 at the Samsung Medical Center in Seoul, Korea, 65 patients, whose bone marrow cells had been stored at initial diagnosis and were available for cytogenetic analysis, were studied. The male-female ratio was 1.24 and all patients were treated according to the Children's Cancer Study Group (CCG) protocol. The median age at diagnosis was five years (range, 3 months-15 yr). The leukemia immunophenotype was determined by standard immunofluorescence analysis using a panel of monoclonal antibodies. Patients were classified into three risk groups by prognostic factors such as age, sex, white blood cell (WBC) count, immunophenotype, and other findings (Table 1). A institutional ethical committee approved this study.
Conventional G-banding analyses
Cell culture and chromosome preparation were performed according to two different protocols: synchronized and unsynchronized techniques in parallel. Synchronization was accomplished using methotrexate. Standard cytogenetic preparations were made. At least 20 metaphases were analyzed using Giemsa-trypsin staining. Karyotypes were interpreted according to the International System for Cytogenetic Nomenclature (ISCN) (11).
Interphase fluorescence in situ hybridization (FISH)
The selected probes were LSI BCR/ABL ES (extra signal) Dual Color Translocation Probe (Vysis Inc., Downers Grove, IL, U.S.A.), LSI TEL/AML1 ES Dual Color Translocation Probe (Vysis), MLL Dual Color Break Apart Rearrangement Probe (Vysis), and LSI p16/centromere enumeration probe (CEP) 9 Dual Color Probe (Vysis). FISH was performed according to the manufacturer's instructions. The image was analyzed using Cytovision (Applied Imaging International Ltd., Newcastle, England). At least 200 interphase nuclei were analyzed for each case, and if needed, the metaphases were also analyzed. Nuclei with ambiguous signals were excluded from analysis.
To determine cut-off values of each gene rearrangement, we analyzed 20 bone marrow specimens from patients without evidence of hematologic malignancies or solid tumors by FISH. Each cut-off was set at mean proportion of cells with rearrangements plus 3× standard deviations. The cut-off values were as follows: 1.5% for BCR/ABL translocation; 1.0% for MLL translocation; 3.4% for MLL deletion; 1.5% for TEL/AML1 translocation; 3.1% for p16 deletion.
RESULTS
Cytogenetic investigations were performed by conventional G-banding analysis for all cases and by interphase FISH using probes to detect BCR/ABL rearrangements (57 cases), TEL/AML1 rearrangements (64 cases), MLL rearrangements (62 cases), and p16 deletions (64 cases). Numerical and/or structural aberrations were identified in 73.8% of all cases by the combination of conventional G-banding and interphase FISH, while abnormalities were detected in 49.2% of the cases using G-banding alone. Gene rearrangements were disclosed by FISH in 24 (72.7%) of 33 patients who showed a normal banded karyotype or no mitotic cell in G-banding. Among 30 cases harboring structural abnormalities identified by FISH, only three cases showed chromosome aberrations suggesting the FISH results in G-banding analysis (two patients with 11q23 abnormalities and one patient with translocation involving 12p12). Twenty-one patients (32.3%) including two who had been classified as low-risk group but later had relapsed, had prognostically unfavorable gene rearrangements that had not been previously detected by G-banding analysis.
The most common gene rearrangement was p16 deletion (20.3%) and the incidences of others were 14.1% for TEL/AML1, 11.3% for MLL, and 1.8% for BCR/ABL translocations (Table 2).
Five cases showed the coexistence of more than two gene rearrangements. Three cases showed p16 deletions in combination with other gene rearrangements: one case had MLL deletion, TEL/AML1 translocation, and p16 hemizygous deletion; one had MLL translocation and p16 hemizygous deletion; and the other had t(8;14) with p16 homozygous deletion. The other two cases had TEL/AML1 fusions accompanied by rearrangements of non-translocated TEL gene. Table 3 describes the patients with gene rearrangements.
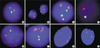
ABL deletion
One case displayed only one ABL signal with two BCR signals in 199 of 200 interphase cells (Fig. 1A). The karyotype was 46,XY,del(5)(q32q34). Considering the presence of both signals from the p16 and CEP9 in the p16/CEP9 FISH analysis, this finding indicated deletion of the ABL gene rather than monosomy 9.
MLL rearrangements
Two different types of MLL gene rearrangements were observed in FISH analysis; translocation and deletion. Six of seven cases with MLL translocations showed no chromosome abnormality involving 11q23 in G-banding analysis. Two cases including one with del(11)(q23) in G-banding, showed only one fusion (orange/green) signal in 19% and 41% of 200 interphase cells, respectively (Fig. 1B). Consequently, a large deletion occurring proximal to the MLL breakpoint was suggested. The possibility of monosomy 11 could be excluded because FISH using the CEP11 confirmed the presence of both chromosome 11.
TEL/AML1 rearrangements
TEL/AML1 translocations were found in nine (14.1%) of 64 patients and none showed t(12;21) in G-banding analysis. Two fusion signals were observed in two cases (Fig. 1C). Two patients (3.1%) with TEL/AML1 fusion had another structural rearrangement involving non-translocated 12p13 breakpoint. One had reciprocal translocation involving 12p12 and 14q21, and the other had deletion of non-translocated TEL gene (Fig. 1D). The proportion of the cells with signals suggesting TEL/AML1 fusion with TEL deletion was 64% of 200 interphase cells. When we performed FISH using the CEP12, 9.0% of 200 cells showed one CEP12 signal and thus we excluded the occurrence of TEL/AML1 fusion in clones with monosomy 12. Four patients showed the loss of one AML1 signal in addition to the TEL/AML1 fusion (Fig. 1E).
AML1 amplification
One case displayed more than five AML1 signals localized in a very defined chromosomal region in 45% of 200 interphase cells, strongly suggestive of intrachromosomal amplification of the AML1 gene (Fig. 1F).
p16 deletions
The combined incidence of hemizygous and/or homozygous deletions determined here was 20.3%. None with p16 deletions had 9p21 abnormalities in G-banding analysis. Homozygous deletions were observed in eight cases (12.5%) and hemizygous deletions in six cases (9.4%). One case had both in two different cell populations. p16 deletions were significantly more common among T-lineage ALL (T-ALL) patients than among precursor-B ALL patients (57.1% vs. 17.2%, p<0.05).
Associations between the gene rearrangements and clinical features or outcomes
Seven patients were lost on follow-up. All patients except four succeeded in attaining complete remission after induction chemotherapy. Five patients died and seven relapsed. Allogeneic bone marrow transplantations were done in nine patients.
Due to the small number of patients in each genetic subgroup and the short duration of follow-up, we did not analyze the relationship between the outcome and each gene rearrangement. However, eight of fourteen patients with unfavorable outcome showed MLL translocations or p16 deletions.
DISCUSSION
We performed FISH with probes for BCR/ABL, MLL, TEL/AML1 rearrangements, and p16 deletions to estimate the incidences of different genetic subgroups with abnormalities involving above genes in Korean childhood ALL, to identify new abnormalities, and to demonstrate the usefulness of FISH. A significant increase in detection rate from 49.2% to 73.8% was observed using the combination of conventional G-banding and interphase FISH analysis. Especially of note, FISH was useful to identify the cryptic gene rearrangements in cases with normal banded karyotype or no mitotic cell in G-banding. The conventional G-banding analysis was able to identify the structural abnormalities in few patients with positive FISH results. Moreover, FISH revealed unfavorable gene rearrangements in two patients who had been treated following the protocol for low or intermediate-risk group and eventually relapsed. Therefore, performing FISH at diagnosis would be important to acquire prognostically important information in childhood ALL.
Compared with incidences of other regions, the incidences of TEL/AML1 translocation and p16 homozygous deletion appeared a little lower in our study. The incidence of TEL/AML1 fusion was 14.1% of total childhood ALL and 15.8% of precursor-B ALL in this study. Incidence of around 25% was reported in the United States (12, 13), Germany (14), Italy (14), and France (15). Although differences in technique, criteria for acceptance, and criteria for inclusion in the studies may account for many of these variations, our results also supported the existence of geographical differences in genetic propensity for TEL/AML1 fusion in childhood ALL (9, 10, 16). The incidence of p16 homozygous deletion was 42.9% in T-ALL and 8.6% in precursor-B ALL, lower than that reported (64% in T-ALL and 23% in precursor-B ALL) (17). However, the higher frequency in T-ALL was concordant with previous report (17).
Frequent rearrangements of non-translocated TEL gene were observed in more than 50% of the patients with TEL/AML1 fusion (8, 15, 18), which supports the theory that the TEL/AML1 fusion gene acts in a recessive manner with regard to TEL gene, or that the secondary genetic changes including rearrangements of non-translocated TEL gene are needed in leukemogenesis by TEL/AML1 fusion (19, 20). However, only two (22.2%) cases among nine with TEL/AML1 fusion showed simultaneous rearrangements of non-translocated TEL gene in our study. This may be due to the relatively low sensitivity of the FISH method. By using molecular genetic methods such as RT-PCR, loss of heterozygosity (LOH) analysis, and spectral karyotyping (SKY), the detection of nontranslocated TEL gene rearrangements will be increased. The design of new FISH probes similar to the MLL break-apart probe could be proposed for simultaneous detection of both the translocation and deletion of non-translocated TEL gene. One patient with non-translocated TEL deletion was classified into the high-risk group, whereas other patients with TEL/AML1 fusion alone were classified into low- or intermediate-risk group in our study. Although the clinical significance of TEL deletion is unclear, this finding suggests a certain role for TEL deletion in the progression of the disease (5, 21). Therefore, investigation for the rearrangements of non-translocated TEL gene in patients with TEL/AML1 fusion will be helpful for predicting the prognosis.
Among nine patients with TEL/AML1 fusions, two showed double fusion signals. One displayed trisomy 21 in G-banding analysis and we assumed that the additional chromosome 21 might be not the normal chromosome but the der(21) t(12;21). It was also reported that the additional fusion signals resulted from duplication of der(21)t(12;21) or ider(21) (q10)t(12;21) (10). Because the TEL/AML1 fusion transcript encodes a strong repressor that interferes with AML1-dependent transcription activation and the wild-type TEL gene, the two fusion transcripts may result in increased expression of the TEL/AML1 fusion gene and increased silencing of the wild-type TEL gene (19, 22). In support of this hypothesis, extra copies of der(21)t(12;21) were found more frequently among patients suffering from relapse (23); our case had deletion of non-translocated TEL.
A large proportion of presumptive del(11)(q23) or del(11) (q23q25) might represent previously unidentified translocations that could be detected by FISH (24, 25). However, our patient with del(11)(q23) in G-banding analysis had also deletion of the MLL gene in FISH. Our two patients with MLL deletions showed relatively longer survival, which was in concordance with the report that MLL deletion was associated with good prognosis (25). As different prognoses between the patients with MLL translocation and those with MLL deletion were reported, FISH would be needed in patients with del(11)(q23) in G-banding analysis.
New findings observed in this study were the deletions of the ABL and AML1 gene. Deletion of ABL was observed in a 13-yr-old boy with T-ALL. Deletions of the 9q34 region on which the ABL gene is located have been known to be quite common findings in several solid tumors (26, 27). However, the deletions of 9q34 have rarely been found in ALL and thus the clinical significance is unknown. Searching for identical cases and additional follow-up study of our case will be helpful in understanding the role of ABL deletion in leukemogenesis of ALL. As well, deletion of AML1 was accompanied with TEL/AML1 fusion and observed in four female patients with precursor-B ALL, which has not yet been reported. It was unclear whether the deletion occurred at der(21) or at normal chromosome 21. The loss of 21q was not observed in patients with TEL/AML1 fusion, while the gain of 21q was frequently found (19). As such, we assumed that the deletions might occur at the der(21) and the AML1 deletion would be one of the secondary genetic changes required in leukemogenesis by TEL/AML1 fusion.
Amplification of the AML1 gene was observed in a 10-yr-old male patient with precursor-B ALL. Since first reported (28), ten patients with AML1 amplification have been subsequently reported (7, 29-32). All cases had childhood precursor-B ALL. One of ten showed TEL/AML1 fusion and none showed TEL deletion. Although six cases were initially classified into the high-risk group, all but one remained alive without relapse. Our case also remained alive without events. AML1 amplification was suggested that play an important role in leukemogenesis as a target event in a trisomy 21 or a 21q22 amplicon (31). Further studies are needed to know the role of amplification of the AML1 gene in leukemogenesis of ALL.
The frequent presence of TEL and p16 rearrangements in the company of other genetic changes suggested that the rearrangements of tumor suppressor genes might contribute to leukemogenesis in cooperation with other genetic changes, possibly by amplifying the malignant potential.
In conclusion, routine performance of interphase FISH using BCR/ABL, MLL, TEL/AML1, and p16 probes at diagnosis would be very useful to establish accurate prognosis and to monitor the minimal residual disease in childhood ALL. Further study with a larger number of patients would be necessary to know the relationship between the outcome and each gene rearrangement and to provide a better understanding of leukemogenesis by new gene rearrangements identified in this study.


 XML Download
XML Download