

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ginseng, the root of Panax ginseng C.A. Meyer, is a well-known folk medicine and has been used as a tonic for over 2000 yr (1-3). Recently there has been a renewed interest in investigating ginseng-pharmacology using biochemical and molecular biology techniques (2).
Pharmacological effects of ginseng have been demonstrated in the central nervous, cardiovascular, endocrine, and immune systems (1-3). In addition, anti-neoplastic, anti-stress, and antioxidant activity have been ascribed to ginseng and its constituents (4, 5). Ginsenosides, which are a diverse group of steroidal saponins and triterpene derivatives containing sugar moieties, are the main active ingredients of ginseng with more than 30 ginsenosides isolated from the roots of Panax ginseng (1, 2).
Recognition that delayed biochemical reactions contribute substantially to tissue damage after head injury has led to the development of targeted neuroprotective strategies in order to limit such secondary posttraumatic cell death and to improve neurological recovery (6).
Previous reports demonstrated the neuroprotective effect of ginsenosides in vitro but rarely in vivo, especially in head trauma (1). In the present study, we evaluated whether treatment with ginseng total saponins (GTSs) can decrease hippocampal neuronal loss, cortical contusion volume, and neurological dysfunction following a controlled cortical impact (CCI) injury.
MATERIALS AND METHODS
Subjects
Twenty-four adult male Sprague-Dawley rats weighing 200-250 g were used for these experiments. All procedures were performed in accordance with the guidelines for care and use of laboratory animals approved by Chung-Ang University's Institutional Animal Care and Use Committee. Ginsenosides used in this study were GTSs obtained from the Korea Tobacco and Ginseng Research Institute (Daejon, Korea). One hundred grams of ginseng was boiled gently in 1,000 mL of water for 60 min. The extract was then concentrated under reduced pressure to obtain a residue.
Surgical procedures
The rats were anesthetized initially with ketamine hydrochloride (15 mg/kg, i.m.) and the head was fixed in a stereotaxic device (Small Animal stereotaxic instrument, David Kopf instrument, Tujunga, CA, U.S.A.), then maintained by 2% halothane mixed with oxygen and compressed air. After a 1.5 cm midline skin incision, a 5 mm diameter craniectomy was made over the right parietal cortex with an electric drill. The craniectomy was centered 3 mm lateral to the sagittal suture and 3 mm posterior to the bregma. Great care was taken to avoid damaging the underlying dura mater during the drilling and removal of the cranial bone.
Experimental controlled cortical injury
Traumatic brain injury (TBI) was performed using the CCI method as described previously (7). After the small craniectomy, an injury was produced using a CCI device (CAUH-2). The device consisted of a 4 mm metal impact tip that was pneumatically driven at a predetermined pneumatic pressure (70 psi), depth (3 mm), and duration of brain deformation (0.2 sec). The penetration depth of 3.0 mm was able to produce a moderate cortical impaction. The wound was closed with 3-0 silk sutures.
Administration of total saponins
The rats subjected to CCI injury were divided into three groups with six rats per group. Intra-peritoneal injections of GTSs or saline were performed immediately after injury (≤3 min post-injury). Rats in the 100 mg-GTSs, and 200 mg-GTSs-treated groups received GTSs dissolved in 1.0 mL saline at a dose of 100 and 200 mg/kg, respectively. Rats in the saline-treated group received 1.0 mL of saline. The sham-operated animals (n=6) received neither GTSs nor saline.
Neurological evaluation
Neurologic evaluation was performed after the TBI using the previously described method (8, 9). The neurobehavioral battery of tests consisted of a rotarod test using the Rota-Rod/7750 (Ugo Basile, Co., Comerio, Italy), beam-balance performance, and posture reflex test.
In the rotarod test, rats were placed on an accelerating rotarod. The time each rat remained on the rod was registered automatically. Neurologic deficit was estimated to be the time at which the rat could no longer remain on the rotarod at a speed of 40 rpm, up to 420 sec. If the rat remained on the rod longer, the test was completed and scored as 420 sec.
Vestibular function was evaluated based on beam-balance performance. Rats were placed on the beam with their head away from the wall, and allowed to remain for 60 sec. Each rat was given three trials, and a mean score was determined.
The posture reflex test evaluated movement asymmetry of the forelimbs as the rat was suspended by the base of the tail. Normal posture was determined to be the symmetrical reaching of both forelimbs toward the ground and was given a score of 3. Moderately abnormal posture, which was the flexion of the contralateral forelimb toward the body, was given score of 2. Severely impaired posture, characterized by flexion of the forelimb and rotation of the shoulder and/or body, was given a score of 1.
Neurological assessments were evaluated 24 hr before and after TBI by an investigator blinded to the study groups.
Extraction of the brain
After neurological evaluation, the rats were anesthetized again with halothane and the brains were removed from the calvarium and weighed. There was an identifiable focal cortical injury at the right parietal cortical area. The brain was cut into 2 mm-thick slices with a rat brain matrix (Harvard Bioscience Co., Holliston, MA, U.S.A.), and stained with 2% 2,3,5-triphenyl tetrazolium chloride (TTC, Sigma, Inc., St. Louis, MO, U.S.A.) at 37℃ for 10 min. After staining, tissue slices were fixed with 4% buffered paraformaldehyde.
Contusion volume
The injury volume was calculated with a digital camera and an image analyzer program (OPTIMAS 6.5, Optimas, Inc., Bothell, WA, U.S.A.).
Histopathological study
The brain slices at the maximum injury site were made into paraffin-blocks. The prepared brain sections were cut into 5 µm-thick slices for hematoxylin and eosin (H-E) staining. Neuronal cells were counted in the hippocampal regions of CA1, CA2, and CA3 by a pathologist blinded to the study groups. For each rat, the mean number of neuronal cells was obtained by examining at least three serial coronal sections in the hippocampus underlying the area of contusion. In each section, neurons were counted in three non-overlapping fields under a high magnification (×400) light microscope in each sub-region (CA1, CA2, and CA3). Only complete neuronal cells with a clearly defined cell body and nucleus were counted.
Apoptosis staining
TUNEL (TdT-mediated dUTP-biotin nick end-labeling) staining was performed using previously described methods (10), and was carried out using the TACS2 TDT-DAB kit (Trevigen, Inc, Gaithersburg, MD, U.S.A.). TUNEL stained cells were counted under a high magnification (×400) light microscope.
Statistical methods
Neurological scores (Table 1), contusion volume (Table 2), and neuronal cell counts (Table 3) were presented as mean scores±SD of the six rats per group. Data was compared between groups using one-way analysis of variance (ANOVA) followed by a Tukey-Kramer multiple-comparisons method. P values of <0.05 were considered statistically significant.
RESULTS
Neurobehavioral assessment
24 hr after CCI injury, injured rats showed significant neurological deficits compared with the sham-operated rats. CCI injury led to definite impairments on all neurological function tests in saline-treated rats compared with the sham-operated rats (Table 1). The results of the rotarod test, beam balance performance test, and posture reflex test for saline-treated rats were 116.5±30.5 sec, 50.0±9.2 sec, and 1.8±0.4, respectively (Table 1). In rats treated with GTSs (100 or 200 mg/kg) after the CCI injury, neurological deficits were improved significantly compared to saline-treated rats (Table 1). Rotarod test results for the 100 and 200 mg-GTSs-treated groups were 165.0±43.7 sec, and 251.2±53.0 sec respectively. The 200 mg-GTSs-treated rats showed a definite improvement in the rotarod test compared to saline-treated rats (p<0.05). Beam-balance performance and posture reflex test showed no significant difference between groups (p>0.05) (Table 1). Sham-operated rats did not show any neurological deficits.
Contusion volume
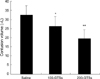
Contusion area was easily defined with 2% TTC staining in all injured rats (Fig. 1). Contusion volumes in the saline-treated group, 100 mg-GTSs, and 200 mg-GTSs-treated group were 32.5±3.5 µL, 26.5±2.9 µL, and 19.5±2.7 µL, respectively. Treatment with GTSs (100 or 200 mg/kg) significantly reduced the contusion volume by 18.5% in the 100 mg-GTSs-treated rats and 40% in the 200 mg-GTSs-treated rats relative to saline-treated rats (Table 2) (p<0.05). The effect of GTSs on contusion volume was increased in a dose-dependent fashion (Fig. 2). Sham-operated rats did not show any cortical lesion under 2% TTC staining.
Histopathological outcome
In the saline-treated rats, CCI injury caused significant neuronal loss after 24 hr in the CA2 (53.4%, p<0.05) and CA3 (34.6%, p<0.05) regions of the hippocampus (Fig. 3A, C) compared to the results from contralateral hippocampal region (Fig. 3B, D) (Table 3). Treating rats with GTSs at a dose of 100 mg/kg showed significant decrease of the TBI-induced neuronal loss in the CA2 (22.7%, p<0.05) and CA3 (24.1%, p<0.05) regions relative to contralateral lesions (Table 3). 200 mg/kg GTSs dosage also showed significant TBI-induced neuronal loss decrease in the CA2 (16.1%, p<0.05) and CA3 (13.9%, p<0.05) regions (Fig. 3E, G) (Table 3). The protective effect of GTSs on hippocampal cell death was increased in a dose-dependent fashion (Table 3). There was no significant difference between groups in the CA1 region (Table 3) (p>0.05).
TUNEL staining
TUNEL-positive cells were found in the pericontusional zone in all injured groups. TUNEL-positive cells were not found in the contralateral cortex or in the contralateral hippocampal area. The number of TUNEL-positive cells was 11.5±1.9 cells/HPF in the saline-treated rats, 10.8±1.5 cells/HPF in the 100 mg-GTSs-treated rats, and 10.3±1.8 cells/HPF in the 200 mg-GTSs-treated rats. There was no significant difference in the number of TUNEL-positive cells between groups (p>0.05). There were no TUNEL-positive cells in the sham-operated group.
DISCUSSION
In general, TBI mechanisms include changes in ionic homeostasis (such as calcium, potassium, sodium, and magnesium), release of excitatory amino acids, induction of free radicals, inflammatory/immune changes, and alterations of multiple neurotransmitter systems (11). It is also known that TBI leads to apoptotic and necrotic cell death, but both cell death forms may be pharmacologically modulated (12).
The CCI model can produce graded, reproducible, and quantifiable brain injury in rats (13-15). The severity of the CCI injury in rats was defined by measuring the pathological effects of varying compression and impact velocity (16). Cortical injury, hippocampal neuron loss, and neurological deficits were found to be produced by moderate-grade CCI injury (15). We created a 3.0 mm deformation by applying a 3.0 m/sec velocity, which corresponds to a moderate grade of injury. We restricted studies to moderate injury because severe-grade CCI produces extensive brain damage resulting in a high mortality rate.
In this study, data demonstrated that the rotarod test was superior to the beam-balance or posture reflex tests in reliably detecting motor dysfunction differences between groups. Our results were in accordance with a previous study (17).
Previous authors had used TTC staining for quantification of infarction volume after focal cerebral ischemia. Furthermore, TTC has been utilized to assess the effectiveness of potential neuroprotective agents (16). Because moderately severe CCI-induced brain injury may result in penetration, causing a tear in the dura and a significant swelling of the brain, the cortical injury after TBI usually results in a complex shape and indistinct border, making contusion volume measurement difficult (16). We made 2 mm-thick brain slices to measure precise injured volume, and a relatively well-demarcated contusion area could be confirmed. This seemed to be an adequate thickness for contusion area definition and the measurement of contusion volume.
GTSs are a mixture of ginsenosides with various ginsenoside ratios as follows: Rb1, 18.26%; Rb2, 9.07%; Rc, 9.65%; Rd, 8.24%; Re, 9.28%; Rf, 3.48%; Rg1, 6.42%; Rg2, 3.63%; Rh1, 4.70%; R0, 3.82%; Ra, 2.91%; and other minor ginsenosides (1). We observed that GTSs effectively improved neurological recovery following moderate CCI injury. Although the molecular mechanisms of neuroprotection in TBI by ginsenosides are not clear, there are several potential mechanisms. It is possible that ginsenosides may act as antioxidants, or free radical scavengers (18). It is well established that lipid peroxidation by free radicals is one of a number of biological mechanisms responsible for secondary injury after traumatic or ischemic injury to the CNS (11).
Ginsenosides may possess the ability to protect neurons from ischemic damage, and were shown to rescue hippocampal neurons from lethal ischemic damage possibly by scavenging free radicals, which are overproduced in situ after brain ischemia and reperfusion (19, 20). Another study described that the antioxidant action of ginsenosides was based on the chemical structure of ginsenoside Rb1 similar to that of 21-aminosteroids (19). It also binds to glucocorticoid receptor with specific affinity and triggers transcriptional activation of glucocorticoid response elements-containing receptor plasmids (21). It promotes cell proliferation and enhances the survival rate of new-born cells (20).
We could not find a difference in the number of TUNEL-positive cells between groups. Previous studies describe that ginsenosides induce (22) or inhibit apoptotic cell death (23). Further studies are needed to evaluate the effect of ginsenosides on apoptosis relating to the TBI.
This study suggests for the first time that ginsenosides show a neuroprotective effect after experimental traumatic brain injury. However, because this study reflected effects of GTSs at 24 hr after traumatic brain injury, we could not define the time of maximal effect and duration of action. Further studies are needed to evaluate the effects of individual ginsenosides in TBI.





 XML Download
XML Download