

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Complement component 7 (C7) is one of the five terminal complement components that generate a multi-protein complex known as the membrane attack complex (MAC) upon activation of either the classical or alternative complement system. MAC is comprised of the complement components C5b, C6, C7, C8, and C9. On cell membranes, this complex becomes MAC, which is capable of forming trans-membrane channels through which ions migrate, leading to cell lysis and cell death (1). The killing function of MAC is best demonstrated in individuals with a homozygous deficiency of MAC protein: since MAC is essential to maintain localization and to prevent dissemination of infection caused by Gram-negative organisms such as Neisseria gonorrhoeae and N. meningitides, these individuals have increased susceptibility to develop a disseminated disease of such infections (2).
The deficiency of C7 leads to recurrent meningococcal infections, and it is often discovered by complement assays in such conditions (3). The C7 deficiency is an autosomal recessive disorder and has mostly been reported in Caucasians (4). The gene encoding the C7 protein has been assigned to the chromosome band 5p13 (5). The molecular bases of C7 deficiencies have been reported, as well as those of subtotal deficiencies of C7 (6-8). In this report, we describe a Korean patient with meningococcal meningitis who was diagnosed as having C7 deficiency based upon complement assays and C7 gene mutation analysis. This is the first confirmed case of C7 deficiency in Korea and the two mutations detected in the patient have not been reported previously.
MATERIALS AND METHODS
Subjects
The proband, an 11-yr-old Korean girl, was admitted because of meningococcal meningitis. Laboratory screening tests for underlying immunodeficiency revealed that the total hemolytic activity (CH50) was undetectable in the serum of the patient. Under the suspicion of an inherited complement deficiency, her family members, both parents and two siblings, were recruited for further studies after obtaining informed consent. The total serum hemolytic activities in these family members were within normal range (Fig. 1). Subsequent analysis by radial immunodiffusion assay with antibodies against the main principal complement components revealed that there was no detectable C7 in the serum of the patient, whereas the C7 concentrations in the sera of both parents and younger brother were 49-52% of the normal plasma level (reference range, 80-120%). The functional deficiency of C7 was confirmed by a hemolytic assay that showed the patient's serum was unable to recover the hemolytic activity of C7-deficient serum. The C6 levels of the patient and the family members were all within normal range.
Direct sequencing of the C7 gene
Genomic DNA was isolated from peripheral blood leukocytes using the Wizard Genomic DNA Purification kit according to the manufacturer's instructions (Promega, Madison, WI, U.S.A.). All coding exons and flanking intron regions of the C7 gene were amplified by using primer sets designed by the authors (available upon request). The polymerase chain reaction (PCR) was performed with a thermal cycler (model 9600, Applied Biosystems, Foster City, CA, U.S.A.) as follows: 32 cycles of denaturation at 94℃ for 30 sec, annealing at 60℃ for 30 sec, and extension at 72℃ for 30 sec. The amplicon (5 µL) was treated with 10 U shrimp alkaline phosphatase and 2 U exonuclease I (USB Corp., Cleveland, OH, U.S.A.) at 37℃ for 15 min and was incubated at 80℃ for 15 min for enzyme inactivation. Cycle sequencing was performed on the ABI Prism 3100 Genetic Analyzer with the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA, U.S.A.).
Paternity testing and haplotype analysis
Paternity testing was performed using 5 short tandem repeat (STR) markers from 5 different chromosomes (D7S820, D8S1179, D18S51, D21S11, and TH01 on chromosome 11p15.5). Haplotype analysis for the C7 gene region was performed using 7 STR markers flanking the C7 gene (D5S630, D5S416, D5S2031, D5S419, D5S1993, D5S674, and D5S426) and 3 intragenic single nucleotide polymorphisms (SNP; c.665G>A, c.1166G>A, and c.1182+10G>A). All STR markers were obtained from the ABI PRISM Linkage Mapping Set v2.5 (Applied Biosystems) and were analyzed on the ABI Prism 3100 Genetic Analyzer and the Genescan software (Applied Biosystems).
Gene dosage analysis
To confirm the deletion mutation of the C7 gene, a multiplex PCR system was designed. The fourth exon of the low-density lipoprotein receptor (LDLR) gene was arbitrarily selected and was simultaneously amplified with each exon of the C7 gene. After electrophoresis of the PCR products on a 2% agarose gel, the density (D) of each band was measured by using the Gel-Doc 1000 Documentation System and Quantity One Software (Bio-Rad Laboratories, Hercules, CA, U.S.A.). Any exon with a density ratio (DR) less than 0.75, an arbitrary cutoff, was considered as being deleted: DR={[patientDC7 exon/(patientDC7 exon+patientDLDLR exon 4)]/[controlDC7 exon/(controlDC7 exon+controlDLDLR exon 4)], where D is the band density of any exon of the C7 gene or exon 4 of the LDLR gene.
RESULTS
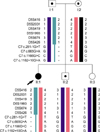
The sequencing analysis indicated that the patient was a homozygous carrier of a G-to-T transversion mutation in the consensus splice-acceptor site of the intron 4 (c.281-1G>T), presumably resulting in skipping of exon 5 of the C7 gene during the RNA processing and splicing step. This led us to suspect that the patient's parents would be heterozygous carriers of this splice-site mutation; however, the sequencing analysis revealed that the patient's father was homozygous for the wild type allele for this locus, while her mother was a heterozygous carrier for the mutation (Fig. 2). In addition, the patient was homozygous for three intragenic SNPs (c.665G>A in exon 7, c.1166G>A in exon 10, and c.1182+10G>A in intron 14), while her father was homozygous for the opposite alleles for the three SNPs.
From this observation, we first performed paternity testing with five STR markers (D7S820, D8S1179, D18S51, D21S11, and TH01), which indicated a paternity probability of 0.999 between the patient and his father (data not shown). Haplotype analysis using 7 STR markers flanking the C7 gene and 3 intragenic SNPs showed that the patient inherited biparental alleles for all STR markers but had only mother's alleles for the c.281-1G>T mutation and all 3 intragenic SNPs (Fig. 3). Therefore, the patient and her father were suspected to have a large deletion mutation encompassing at least from the splice-site mutation to intron 14 of the C7 gene. Further analysis by a semi-quantitative gene dosage analysis using the multiplex PCR method showed that all exons of the C7 gene except for exon 18 were deleted (Fig. 4). To delineate the telomeric breakpoint of the deletion, three exons of the CARD6 gene, located immediately telomeric to C7, were tested and were shown to have a normal gene dosage, indicating that the breakpoint should lie between the CARD6 gene and the C7 gene, whereas the centromeric breakpoint occurred between exon 17 and exon 18 of the C7 gene (standard nomenclature: c.1-?_2350+?del).
The younger brother of the patient inherited the c.281-1G>T mutation from his mother. All heterozygous carriers (the patient's father, mother, and younger brother) showed decreased C7 levels and functions but had normal levels of CH50.
DISCUSSION
C7 is a single-chain polypeptide composed of 821 amino acid residues and is structurally similar to the other components of MAC; C6, C8α, C8β, and C9 (9, 10). The C7 gene spans about 80 kb of DNA and is encoded by 18 exons (10). To date, more than 15 different molecular defects leading to total or subtotal C7 deficiency have been reported (11, 12). Here we report two novel mutations of the gene in a Korean family with C7 deficiency. In order to identify mutations in the C7, we performed a sequencing analysis for all exons and their flanking introns of the gene. The patient was homozygous carriers of a splicing mutation (c.281-1G>T) and three polymorphisms of the C7 (c.665G>A, c.1166G>A, and c.1182+10G>A). As expected, the patient's mother was a heterozygous carrier of the mutation and all three polymorphisms, but the patient's father had wild type alleles for all these variations. Extended haplotype analysis with 7 STR markers flanking the C7 revealed that the patient inherited a large deletion of the C7 from her father. Gene dosage analysis by multiplex PCR of each exon showed that the deletion encompassed from exon 1 to exon 17, the largest deletion reported to date in the terminal complement deficiency. The G-to-T transversion mutation in intron 4 generates a defect in the splice acceptor site and results in skipping of exon 5 (148 bp), which is expected to produce a premature stop codon at position 130. These mutations have led to a loss of complement lytic function. The family study demonstrated that the patient's brother also carried one C7 allele with the G-to-T transversion mutation in intron 4. Nonsense or frameshift mutations in human disease genes frequently cause severe reduction of corresponding mRNA and resultant protein deficiency. Occasionally, nonsense mutations are associated with normal mRNA levels, but truncated proteins. The mechanisms for the reduced expression of nonsense mutated mRNA are incompletely understood, but they seem to be diverse and to involve cytoplasmic and nuclear rates of decay of cytoplasmic mRNA (13, 14).
Epidemiological data on terminal complement deficiencies show that they, as a group, are more prevalent than early component deficiencies. In addition, there appears to be an ethnic predilection for specific terminal complement deficiencies: C6 and C8α-γ deficiencies are common in blacks, whereas C7 and C8β deficiencies are predominantly observed in Caucasians. The cases of C7 deficiency have been reported more frequently in Spanish, Israeli, and Irish than in Asians in Japan and Korea (6, 7, 11, 12).
Of interest, there is no uniform association between any specific geographic/ethnic group and meningococcal disease. While there was no patient with a complement gene deficiency among the survivors of Neisserial epidemics in the Faroe Islands and Denmark (15, 16), a high prevalence of C7 deficiency was noted among Moroccan Jews in Israeli populations (17, 18). These data may reflect environmental factors such as the endemic rate of infections and climatic differences and/or genetic differences in the host population (19). An ancillary finding in many studies of families with complement-deficient propositus is the existence of complement-deficient siblings who had never been infected. This also indicates different susceptibility among complement-deficient individuals with the same genetic defect, e.g., C6 deficient patients who had survived one episode of meningococcal infection or had never experienced an infection were shown to be at less risk than those who had experienced recurrent infections (20). On the other hand, the immunity to meningococcal disease may also be determined by the interaction between IgG and the IgG Fc receptor (FcγR) on polymorphonuclear cells (antibody-mediated phagocytosis). It has been hypothesized that particular combinations of FcγR allotypes may determine the susceptibility to meningococcal disease, especially in persons who lack serum bactericidal activity because of deficiency of a component in the terminal pathway of the complement system (21, 22). Recent study by van der Pol et al. found that there was no statistical significance in the association between particular combinations of FcγR allotypes and meningococcal disease (23). These authors suggested that the genetic heterogeneity on the long arm of chromosome 1, which is enriched with immune-modulating genes, including pentraxin, selectin genes, and several complement regulatory proteins, may provide important information with respect to host defense against meningococci (24).
Screening for complement deficiencies is not a priority in most countries. Routine screening of a child after meningococcal disease is not necessary, if the infection is caused by group B or C. However, an investigation is warranted if there is a relevant past medical history such as previous suspicious infections, an abnormal course of infective illness, infection with non-groupable or unusual serogroups or if the current illness is a repeated episode of neisserial infections (25). Administration of prophylactic antibiotics should be considered in a patient with complement deficiency, although it may encourage drug resistance (20). Also, vaccination with the tetravalent polysaccharide vaccine can prevent meningococcal disease due to the meningococcal serogroups A, C, Y, and W135 (26).



 XML Download
XML Download