

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is an important differential diagnosis in infants and children who present with prolonged fever, pancytopenia, splenomegaly, marked triglyceridemia, and systemic hypercytokinemia without evidence of aplastic anemia or malignancy (1-4). In this disorder of the mononuclear phagocytic system, infiltrating lymphocytes and histiocytes destroy the central nervous system (CNS), bone marrow, and other organs (1). HLH occurs in both primary (familial) and secondary (infection- or malignancy-associated) forms (1-5). Although fatal cases occur most often in patients with the familial form (FHL) rather than the virus-associated form (VAHS), Epstein Barr virus-associated HLH (EBV-HLH) is implicated in the pathogenesis of fatal VAHS (1, 6-8).
The pathogenesis of EBV-HLH remains poorly understood, although the findings of several recent studies support the concept that the monoclonal proliferation of EBV-infected T cells may be a primary feature of EBV-HLH (9-11). The more common explanation is that EBV-infected B cells trigger polyclonal proliferation of cytotoxic T cells, which in turn stimulate histiocytes and macrophages (1). However, EBV may also target T cells directly in EBV-HLH (1, 10). Most EBV-infected cells are CD8+ T cells in EBV-HLH and CD4+ T cells or NK cells in chronic active EBV infection; in contrast, most EBV-infected cells are B cells in acute infectious mononucleosis (12).
The HLH Study Group of the Histiocyte Society and other study groups have long sought to improve the clinical outcome of FHS and VAHS by combining established induction regimens such as epipodophyllotoxins, intrathecal methotrexate, and steroids, with newer forms of immunomodulatory therapy to maintain remission until an acceptable marrow donor is found (2, 3). We now report the results of immunochemotherapy in four patients with EBV-HLH treated from 2001 to 2004 at Chungbuk National University Hospital in Korea.
MATERIALS AND METHODS
Eligibility and diagnostic criteria
Children were eligible to participate if they met the diagnostic criteria for HLH with a positive EBV infection and had not received treatment for this disease. The diagnostic criteria were: fever persisting beyond seven days with peak temperature ≥38.5℃, splenomegaly, cytopenia affecting at least two lineages in the peripheral blood not caused by a hypocellular or dysplastic bone marrow, and hypertriglyceridemia (fasting triglycerides >3 SD or 177 mg/dL) or hypofibrinogenemia (fibrinogen <3 SD or 150 mg/dL) that raised strong suspicion of hemophagocytic syndrome (2, 3). Bone marrow aspiration was performed in all patients and a liver biopsy was performed in one patient (Patient 3) for cytomorphological analysis.
EBV virologic studies
EBV infection was demonstrated by serology, polymerase chain reaction (PCR), and in situ hybridization.
Serological tests for EBV infection were based on viral capsid antigen (VCA)-IgG/IgM, early antigen (EA)-DR-IgG/IgM and Epstein-Barr virus nuclear antigen (EBNA) antibody titers.
Bone marrow cells of patients were examined for the presence of EBV DNA by PCR. DNA was extracted from bone marrow cells with 100 µL of 5% Chelax resin (Bio-Rad Laboratories, Hercules, CA, U.S.A.) and two cycles of heating (up to 100℃) and cooling. Five microliters of the DNA eluate was used for amplification of EBV DNA, with 250 nM concentration of sense and antisense primers (5'-TGATAACCATGGACGAGGAC-3' and 5'-GCAGCCAATGCAACTTGGAC-3', respectively) that correspond to the 139 base-pair fragment of the Epstein Barr Nuclear Antigen-1 (EBNA-1), and Accupower® PCR PreMix (Bioneer, Daejoen, Korea). PCR was performed using 5 min of denaturation at 95℃, 35 cycles of 1 min at 94℃, 1.5 min at 57℃, 2 min at 72℃, and finally 10 min at 72℃. The PCR products were analyzed by electrophoresis using 2% agarose gels. DNA from EBV-transformed human B-cell lines (IM-9) and human T-leukemic cell lines (Molt-4) were used as positive and negative controls, respectively.
In situ hybridization of EBV-encoded small RNA (EBER) was performed according to a modification of a previously described cDNA-mRNA in situ hybridization method, using the manual Microbe System (Fisher Scientific, Pittsburgh, PA, U.S.A.) (13). Briefly, 5 µm thick bone marrow sections were placed on the distal one-third of Probe-On positively charged slides. The specimens were deparaffinized with Hemode (xylene substitute) at a ratio of 3:1 for 5 min, placed in 100% alcohol for 45 sec, and rehydrated with 1 Automation Buffer (Biomeda, Foster City, CA, U.S.A.). The specimens were digested with pepsin (pH 2.0) for 6 min at 110℃ and immediately washed in distilled water for 30 sec. The biotinylated cDNA oligonucleotide probe (Research Genetics, Huntsville, AL, U.S.A.) to EBER-mRNA was reconstituted to 1 µg/µL solution in 10 mM Tris (pH 7.5), diluted to a concentration of 200 ng/mL in the probe cocktail containing 12.5% (v/v) formamide, 0.5% (w/v) chondroitin sulfate, 675 mM sodium chloride, 138 mM sodium citratem, 12 mM sodium phosphate monobasic, 63 mM sodium dibasic, 0.1% (v/v) Tween 20 (JT Baker, Phillipsburg, NJ, U.S.A.) and 1:100,000 diluted random 9-mers (Research Genetics). This probe mixture was applied for 10 min at 92℃ and 30 min at 45℃ to hybridize with target nucleic acids in the specimens. After hybridization, the specimens were washed in 5×SSC for 3 min and 2×SSC for 3 min at room temperature. Detection of hybridized biotin probes was performed with avidin-alkaline phosphatase (Dako, Carpenteria, CA, U.S.A.), diluted 1:200 in Tris buffer (pH 8.0), and signal localization was obtained with the naphthol AS-MX phosphate/Fast Red TR chromogen solution. The slides were washed with distilled water, counterstained with hematoxylin for 5 sec, and coverslips applied with Universal Mount (Research Genetics).
Treatment regimens
The core elements of induction chemotherapy were steroids and etoposide (VP-16). The starting doses were 10 mg/m2 per day oral dexamethasone and 150 mg/m2 per dose intravenous etoposide twice a week. Remission induction therapy, either closely or entirely, followed the HLH-94 protocol in all patients (1-3).
The aim of continuation treatment is to sustain resolution of the disease (2, 3). After eight weeks of induction chemotherapy, a complete response (CR) was defined as an unequivocal resolution of clinical signs and symptoms and the normalization of laboratory findings, including data obtained from imaging studies and resolution of hemophagocytosis on bone marrow study. Patients with persistent fever and other symptoms of HLH, abnormally high serum ferritin concentration or remaining hemophagocytosis on bone marrow study were considered to have a partial response (PR) and were placed on intensified or maintenance regimens until CR was induced. The continuation therapy from week 9 comprised 10 mg/m2 dexamethasone pulses for three days every second week and 150 mg/m2 etoposide infusions on alternating weeks in combination with daily oral cyclosporin (CSA) (1-3).
RESULTS
Characteristics of patients and laboratory findings
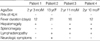
The mean age at diagnosis was 5.2 yr (range 2.3-13.0); two patients were boys and two were girls. The patients had prolonged fever, splenomegaly, hepatomegaly, lymphadenopathy and/or neurological symptoms with cytopenia. None of the patients had a family history of HLH (Table 1).
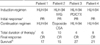
At the time of diagnosis, the serum ferritin concentrations were 364 ng/mL-24,295 ng/mL (normal range 15-332 ng/mL) and the serum lactate dehydrogenase levels were 2,357 IU/L-5,540 IU/L (normal range 180-460 IU/L) (Table 2). Serological testing for EBV showed an elevated VCA-IgG (normal range 0.9-1.09 U/mL), with increased EA-IgG titers (normal range 0.9-1.09 U/mL) indicating reactivated EBV infections in all patients (Table 3). EBV DNA was detected by PCR in Patient 1 and 4 (Fig. 1) and EBER was detected by in situ hybridization in bone marrow specimens of the three patients (Fig. 2). Hemophagocytosis was shown in the bone marrow aspirates of all patients (Fig. 3) and liver biopsy specimen of Patient 3 (data not shown). There were no chromosomal anomalies of peripheral blood mononuclear cells or bone marrow cells (data not shown).
Clinical courses and treatment response
During the first eight weeks of therapy, the four patients exhibited PR and were placed on continuation treatment. Patients were provided therapy for a median of seven months (range 4-10 months) (Table 4).
On hospital day 2, Patient 1 suffered a pulmonary hemorrhage requiring ventilator care; immunochemotherapy by HLH-94 and continuous CSA infusion were started. Severe neutropenia (absolute neutrophil count 48/µL) without evidence of bacterial infection during remission induction led to temporary withdrawal of all cytoreductive therapy and continuous infusions of CSA for 11 days. On hospital day 27, the patient achieved hematopoietic recovery and was able to restart his scheduled chemotherapy. However, this patient was considered to have a PR after induction treatment, because serum ferritin concentration was 374 ng/mL, which was above the normal range. After continuation treatment, the patient achieved CR and his serum ferritin concentration normalized.
Patient 2 had a seizure and loss of consciousness at admission. She presented with neurological deterioration and a brain MRI showed increased signal in the head of the caudate nucleus, medial portion of the thalamus, right lentiform nucleus, cortex and subcortical white matter of the right temporal lobe, and periaqueductal gray matter. Tests for serum antinuclear antibody and anti-double-stranded DNA were negative. She received medication including antibiotics, antiviral agents, and antifungal agents. On hospital day 2, pancytopenia developed and her bone marrow aspirate was characterized by infiltration of hemophagocytosis. Immunochemotherapeutic agents including dexamethasone, etoposide, CSA, and intrathecal methotrexate were administered immediately and her neurological symptoms improved. However, all immunochemotherapy except dexamethasone was stopped because of severe bone marrow suppression. Six days after the cessation of immunochemotherapy, cranial nerve VI palsy occurred suddenly, but it was treated successfully with etoposide and high dose dexamethasone. There was no hemophagocytosis in the bone marrow after one month of chemotherapy. Abnormal brain imaging remained after induction therapy, and she was treated by continuation therapy with HLH-94 protocol. The brain MRI abnormalities resolved after continuation treatment for 10 months. Dexamethasone was discontinued because of progressive abdominal striae and she showed poor compliance to oral CSA. Although CR was attained, she dropped out of the follow-up 20 months after the onset of disease.
Patient 3 received prednisolone and cyclophosphamide and switched to induction treatment by the HLH-94 regimen from hospital day 2. His liver biopsy demonstrated some histiocytic infiltration in the liver sinusoid. Although the patient had shown persistent hepatomegaly over five months, hemophagocytosis subsided after induction treatment.
Patient 4 was admitted with prolonged fever, hepatosplenomegaly, leukopenia, and thrombocytopenia. Immunochemotherapy by HLH-94 from hospital day 5 appeared to successfully induce CR. However, at the end of induction treatment, she presented with recurrent fever, hepatosplenomegaly, and pancytopenia; hemophagocytic histiocytes were noted again in bone marrow, indicating reactivation. The induction treatment was restarted and clinical symptoms and hemophagocytosis of bone marrow disappeared after one month of treatment, after which she was maintained on continuation treatment with no evidence of reactivation thereafter.
DISCUSSION
EBV-HLH poses unusual challenges to pediatric hematologists. The reactive disorder may be difficult to distinguish from infectious mononucleosis, septicemia, certain hematological malignancies, and systemic autoimmune disorders (1). Patients in our study met most of the HLH diagnostic criteria proposed by Henter et al. and lacked a family history of HLH (2-4). It is sometimes difficult to make a clear distinction between FHL and VAHS (8, 14). We believe that our clinical and laboratory findings support the diagnosis of EBV-HLH in our patients. Our diagnosis was based on serological evidence of initial or previous exposure to EBV (e.g., elevated VCA-IgM or IgG) or reactivation (e.g., elevated EADR-IgG) and molecular evidence such as detection of EBV genomic DNA by PCR and EBER by in situ hybridization in bone marrow cells (9, 15). We suggest that sometimes therapy should be started when there is a strong clinical suspicion of HLH, although the patient may not necessarily fulfill all the diagnostic criteria.
The HLH Study Group began its first international treatment strategy dedicated to HLH using HLH-94 in January 1995 (3). Many studies since then have recommended similar therapeutic approaches to newly diagnosed cases of EBV-HLH (1-3). Steroids and etoposide are common treatment agents in both the induction phase and the continuation phase for patients who show resistance to other agents (1-3). Early introduction of an etoposide-based regimen is purported to be a critical factor in securing long-term survival in patients with EBV-HLH (16-18). However, increased incidence of acute myeloid leukemia and myelodysplastic syndrome can occur following the use of etoposide (2, 19). In children with obscure CNS symptoms and a progressive encephalopathy, HLH should be considered as the differential diagnosis because the inflammatory meningoencephalopathy in HLH may cause severe and permanent CNS dysfunction (20, 21). Systemic therapy including dexamethasone, which penetrates the blood-brain barrier well, is first line therapy in HLH-94 protocol (3, 21). Because it has not yet been determined whether intrathecal therapy in combination with systemic dexamethasone is beneficial, the HLH-94 protocol reserves intrathecal therapy for those with clinical evidence of CNS progression or unimproved CSF pleocytosis (2, 3).
A prompt, short-term CSA infusion is recommended during neutropenic episodes. CSA, which has not been widely used in therapy for EBV-HLH, seems to control the deregulated release of cytokines and their receptors in patients with neutropenia (1, 22). Improved neutrophil recovery caused by CSA treatment makes it possible to continue immunochemotherapy safely and to obtain better outcomes (18, 23). CSA can now be added to induction treatment in the HLH-2004 protocol by the HLH Study Group. Imashuku et al. suggested that the choice of treatment should be based on the risk factors, and that CSA, steroids, or intravenous immunoglobulin (IVIG) may be indicated as initial treatment for low-risk patients and etoposide-containing regimens for high-risk patients (23). Although the value of IVIG for induction therapy is controversial (1, 20, 25), IVIG infusion is recommended once every four weeks as supportive therapy in the HLH-2004 protocol. It is controversial whether granulocyte colony-stimulating factor (G-CSF) promotes hematological recovery without disease progression (25, 26).
It is now possible to control a high proportion of EBV-HLH cases without resorting to bone marrow transplantation (BMT) (27). Imashuku et al. reported that 13 of their 17 EBV-HLH patients needed continuation treatment with etoposide and steroids with or without CSA or other forms of chemotherapy; BMT was performed in only two patients who showed resistance to all available immunochemotherapy (1). Allogenic BMT is associated with high morbidity and should be held in reserve and used only for patients with highly continuous refractory disease (1-3). One study reported that 20 of 23 children with nonfamilial HLH were alive and off therapy for more than 12 months without BMT and that most cases (12 of 20 or 60%) were from East Asia (2). Imashuku et al. reported a 78.3±6.7% four-year survival outcome when most patients (85%) were treated with etoposide-containing regimens such as the HLH-94 protocol (16, 28). All of our patients treated with HLH-94 immunochemotherapy are alive without evidence of disease 15 to 27 months (median, 19 months) after treatment. None of our patients was considered refractory to chemotherapy and none required BMT. We suggest that EBV-HLH can be cured with early introduction of immunochemotherapy based on HLH-94 protocol.





 XML Download
XML Download