

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Familial hypokalemic periodic paralysis (HOPP) is an autosomal dominant disorder characterized by episodic attacks of muscle weakness with concomitant hypokalemia (<3.5 mEq/L), which usually involves the four limbs. The age at onset of paralytic crises is usually within the first or second decade. The frequency of attacks is maximal between 15 and 35 yr of age and then decreases with age (1-3). The precipitating factors include carbohydrate- or sodium-rich meals, emotional stress, and rest after exercise. The interval between crisis can vary, and may be prolonged with preventive measures, including avoidance of triggering factors, dietary changes, and appropriate medications.
Recent molecular genetic analysis has shown that HOPP is caused by mutations in a calcium channel gene (CACNA1S) or a sodium channel gene (SCN4A) (2-7). However, the majority of familial HOPP patients have mutations in the CACNA1S gene located on chromosome 1q31-32 (8, 9). The CACNA1S gene encodes the α1-subunit of a skeletal muscle voltage-gated L-type calcium channel known as the dihydropyridine (DHP) receptor. The phenotypes of each mutation have some differences in the gender penetrance, and the clinical characteristics including the response to medications (4, 9-13).
We describe a patient with a mutation of arginine 1239-to-glycine (R1239G) in calcium channel which is the rarest one in the reported familial HOPP patients. In Korea, several cases of familial HOPP in children and adults have been reported (14, 15), but DNA diagnosis for familial HOPP is sparse (16).
CASE REPORT
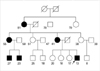
A 13-yr old boy presented with near-daily attacks of mild to severe paralysis for the past 8 yr. The symptoms mainly appeared in the morning and he usually awakened with weakness of the four limbs to the point of difficulty in sitting and walking. After the paralytic episode in the morning, he was free of symptoms in the afternoon, but eating high-sodium or carbohydrate foods and rest after vigorous exercise made him weak or completely paralyzed again at any time. In family history, his mother (41 yr old), maternal-grandmother (81 yr old), maternal aunt (55 yr old) and her two sons (the patient's cousins, 23 and 27 yr old) were also suffering from similar attacks of weakness or paralysis throughout their lives (Fig. 1). The severity and frequency of attacks of grandmother and mother has decreased with age. Two cousins of the proband were diagnosed HOPP 8 yr ago at our hospital. They were treated with acetazolamide and potassium supplements. Because there is no effect of such treatment, they stopped taking the drugs and they were lost further medical support.
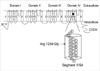
On admission, the proband showed non-specific findings in physical examination, radiologic and laboratory findings including muscle enzymes. During paralytic episodes, the patient showed low serum potassium levels (3.2 mEq/L vs. 4.4 mEq/L between attack) and hypokalemic ECG findings, such as ST segment depression and U-wave. There was no myotonia in EMG findings. DNA examination identified a nucleotide 3705 C to G mutation in exon 30 of CACNA1S gene (Table 1). This mutation predicts codon change from arginine to glycine at amino acid position #1239 of axon 30 in the calcium channel gene (Fig. 2). The patient was treated with spironolactone (25 mg b.i.d.), and he showed a dramatic improvement in his symptoms within 2 days. Other affected members of the family, including two cousins of the proband, have also started taking spironolactone. They could also themselves awaken refresh and move at their will throughout the day.
DISCUSSION
Periodic paralyses constitute a group of human hereditary muscles disorders. Based on the variations of serum potassium levels during attacks, they can classify as HOPP or hyperkalemic periodic paralysis. Both are inherited via autosomal dominant mode with a high penetrance. HOPP is the most frequent cause of periodic paralysis. The prevalence of HOPP has been estimated at 1/100,000. Although most individuals diagnosed with HOPP in Western countries were familial types, sporadic types with or without underlying diseases, such as thyrotoxicosis and renal tubular acidosis have been reported in Korea (14-16).
Three mutations of CACNL1A3 gene are known in familial HOPP (7-9). CACNA1S encodes the α1-subunit of a skeletal muscle voltage-gated calcium channel termed the dihydropyridine (DHP) receptor. This channel is composed of five subunits called α1, α2, β, γ, and δ. The α1-subunit constitutes the ion-conducting pore, and contains the receptor for dihydropyridine and other ligands (17, 18), whereas the other subunits regulate the function of the α1-subunit of the channel. The α1-subunit is composed of four transmembrane domains (I to IV), each of them containing six transmembrane α helices (S1 to S6) (Fig. 2). The amino-acid substitutions in the three mutations are as follows: the replacement of a positively-charged arginine in position 528 in segment S4 of domain II by a weakly-positive histidine (R528H) and an arginine in position 1239 in the S4 segment of the fourth domain by either an histidine or a glycine (R1239H and R1239G) (7, 9). Molecular studies in Caucasian families demonstrated that R528H and R1239H are predominant mutations (4, 10), whereas the R1239G mutation was only found in a single family (9). The molecular diagnosis for familial HOPP was possible using PCR amplication of genomic DNA followed by restriction-enzyme digestion. By this method, the proband identified that the rare R1239G mutation inherited in his family (Fig. 2, Table 1).
Recently, mutations in the sodium channel gene (SCN4A) in HOPP were reported. The structure of the sodium channel is similar to that of the calcium channel and is composed of 4 domains each with 6 segments. These mutations were found in the domain II of the S4 segment like R528H in the mutations of the calcium channel (4, 6).
There are several studies for clinical comparisons in families with different mutations (4, 10-12). Elbaz et al. (10) reported that the R528H and the R1239H mutations had the similar mean age of onset, the number of acute attacks, and the precipitating factors. However incomplete penetrance was observed only in female patients with R528 mutation. Our patients appeared to present severe symptoms in males, and an earlier beginning and higher frequency of paralysis symptoms than the previously reported cases. The response to medications also seems to depend upon the mutations underlying periodic paralysis (4, 13). The acetazolamide, a carbonic anhydrase inhibitor, has been considered highly effective in most HOPP patients (19-21). However, several patients with the R672G SCNA4 mutation (4) and a patient with the R672S SCNA4 mutation (13) showed an exacerbation of the symptoms after acetazolamide treatment. Although we did not perform DNA analysis for two cousins of the proband, they experienced an exacerbation of symptoms with acetazolamide treatment 8 yr ago. However, it has not been determined yet that the R1239G mutation is associated with acetazolamide-resistance. Our patients showed a remarkable improvement of symptoms with spironolactone treatment. Spironolactone and its derivatives are alternatives to acetazolamide if it is not effective after the empirical initial use (22-26). The action of the aldosterone antagonists on HOPP is not known. The beneficial effects on HOPP cannot be explained by antagonism to the action of aldosterone on the renal or enteric excretion of sodium and potassium (22). Although recent studies have demonstrated that mutations in the calcium- and sodium channels cause HOPP, the mechanism of these channels in the pathophysiology of HOPP has not clearly been defined.
Several complications during the clinical courses have been reported. Vascular myopathy, unexpected acute paralysis or cardiac arrhythmia with hypokalemia, and possibly malignant hyperthermia associated with anesthesia have been reported (27, 28).
In conclusion, genetic diagnosis of familial HOPP is very useful for making a confirmative diagnosis, providing genetic counseling for the family members and as well as for providing the appropriate treatment.

 XML Download
XML Download