

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Transverse myelitis (TM) is a rare neurological complication in patients with systemic lupus erythematosus (SLE) (1-3). Moreover TM involving the entire spinal cord longitudinally has only been reported in only a few SLE cases (4, 5). In particular, the medulla oblongata involvement was reported in two cases only (6, 7). Internuclear ophthalmoplegia (INO) presents either as an isolated condition or as one associated with other neurological symptoms (8). INO is an uncommon neurological complication in SLE, and several cases of INO associated with SLE have been observed (9). In particular, the case of TM longitudinally involved accompanied by bilateral INO has never been reported in patients with SLE. Steroid therapy seems effective in improving INO (9), but the outcome of TM involving the entire spinal cord longitudinally is unfavorable despite an aggressive treatment with high dose of steroid and cyclophosphamide in several cases (4, 5). Here, we describe a patient who was diagnosed as having catastrophic-onset longitudinal myelitis involving the entire spinal cord accompanied by bilateral INO associated with systemic lupus erythematosus.
CASE REPORT
A 28-yr-old woman was hospitalized due to a cough, coryza, fatigue, a mild headache, and low-grade fever for three days. Four years ago, she was diagnosed as type IV lupus nephritis by renal biopsy, and had been treated with methylprednisolone pulses and monthly intravenous (i.v.) cyclophophamide pulses for two years. She experienced complete remission and had subsequently been maintained with 300 mg of hydroxychloroquine and 7.5 mg of prednisolone daily. On admission, her vital signs and neurologic examination were normal. Laboratory data showed a white blood cell count of 3,200/µL, hemoglobin of 10.1 g/dL, and a platelet count of 235,000/µL. Her erythrocyte sedimentation rate (ESR) was 80 mm/hr (normal <20). Urine analysis, serum electrolytes and creatinine, liver and thyroid function test, prothrombin time, and activated partial prothrombin time were normal. Her antinuclear antibody (ANA) titer was 1:320 (homogenous pattern) and anti-double-stranded DNA antibody level was over 107 IU/mL (normal 0-7). Serum complement levels were C3 of 27.2 mg/dL (normal 65-125) and C4 of 7.23 mg/dL (normal 12-43), which were lower than those of previous tests. Anticardiolipin antibodies, lupus anticoagulant, anti SSA/SSB antibodies, anti Sm and RNP antibodies, and the Venereal Disease Research Laboratory (VDRL) test were all negative. Plain radiography examination revealed pansinusitis of both maxillary and fronto-ethmoidal sinuses. She was initially treated with antibiotics and 20 mg of prednisolone per day under the diagnosis of sinusitis and flares of lupus.
On the third day of the admission, her previous symptoms became aggravated and she developed a severe headache, nausea, and vomiting. Aspirated cerebrospinal fluid (CSF) contained leukocytes of 54/HPF (neutrophils 87%, lymphocytes 13%), protein of 49.1 mg/dL (15-45), and glucose of 31 mg/dL (50-75). Subsequently, the Gram stain and culture of CSF were found to be negative and aseptic meningitis was diagnosed.
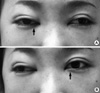
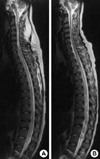
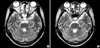
On the fourth day of the admission, the patient presented with an abrupt onset of nystagmus, horizontal diplopia, and mild tingling sensation of the anterolateral aspect of the left thigh. An ophthalmic examination showed medial gaze disturbance of the both eyeballs (Fig. 1), but reduction of visual power, visual field defect, and optic disc abnormality were not detected. We observed bilateral adduction weakness and concomitant abducting nystagmus with intact vertical eyeball movement. Thus, she was diagnosed as having bilateral INO. In addition, a weakness of both legs and voiding difficulty developed. The sensory deficit was consistent with below T10 level, and bladder catheterization yielded 800 mL of residual urine. After 12 hr, flaccid paralysis developed in both upper and lower extremities. The sensory deficit was consistent with below T4 level, and bladder and colon paralysis persisted. An MRI scan of the brain and whole spine showed a diffuse swelling and high signal intensities involving the whole spinal cord, pons, and medulla oblongata (Fig. 2A). Bilateral high signal intensities in T2-weighted image were pronounced in the lower midbrain, pons, medulla oblongata, and periaqueductal region (Fig. 3A). With a diagnosis of acute catastrophic-onset longitudinal myelitis, aggressive treatment with i.v. pulsed methylprednisolone was started immediately. Nonetheless, vigorous dyspnea occurred several hours later and mechanical respiratory support was performed due to respiratory failure and stuporous mental state. Intravenous pulsed methylprednisolone was given for 5 days and i.v. pulsed cyclophosphamide was added in the intensive care unit. Her mental status and respiratory function were slowly improved and returned to normal.
On the ninth day of the admission, motor and sensory functions of the upper extremities were restored to the T10 level, and motor function of both eyeballs was recovered. However, the neurological dysfunction of the lower limbs, and bladder and colon paralysis were almost unchanged. Follow up MR images, which were taken one month later, showed decreased high signal intensity of the cervical spinal cord and brainstem. However, remnant and worsened abnormal changes in the thoracic spinal cord extending to the conus medullaris were identified (Fig. 2B, 3B). The patient was discharged to a rehabilitation clinic and six month later her neurological deficit was unchanged despite monthly i.v. pulsed cyclophosphamide and 20 mg prednisolone a day.
DISCUSSION
TM is a rare neurological manifestation, but causes serious complication in patients with SLE (1-3). Previous reports have indicated about 1-2% prevalence. (10). Moreover, a small number of reports concern MRI-depicted longitudinal myelitis of the entire or the greater part of the spinal cord in SLE (4, 5). Deodhar et al. (11) reported the case of a patient with continuous involvement of the spinal cord from C3 to T2 and from T7 to the conus medullaris. This is the first report of a longitudinal myelitis. Especially, two cases of medulla oblongata involvement have been reported (6, 7). In the case of our patient, the TM extended from the midbrain to the conus medullaris, which has never been reported previously.
This patient's neurological symptoms showed an abrupt catastrophic onset, and rapidly progressed to respiratory failure within several days. TM with an acute catastrophic onset is rarer than acute or subacute onset in SLE (2, 3). Study of Ropper and Poskanzer (3) suggested that three distinct types of onset occur in TM; the largest group has ascending paresthesia at onset, usually with a course that evolves over one to fourteen days; the smaller group has an evolving syndrome over ten days to four weeks with a stuttering course; another group coming on suddenly and evolving over several hours. In particular, the last group showing hyperacute and catastrophic onset followed by the development of a 'spinal shock syndrome' tends to have a poor prognosis. Thus, our patient's case represents the most severe form of the disease.
One of the focal neurological manifestations of SLE is INO. It is defined as a lesion of the medial longitudinal fascicle, which connects the nucleus of the VI cranial nerve pair with the contralateral III pair during horizontal movement (8). INO presents almost invariably during periods of SLE activity and is either isolated or, more often, is associated with other neurological symptoms. However, its prevalence in SLE is low and only a few cases of bilateral INO have been reported (9). This is the first case in the literature of bilateral INO accompanied by TM. MRI has shown brainstem lesions in only two of the literature-reported INO patients (12, 13). Our patient's MR images showed definite brainstem lesions, which were deeply associated with internuclear ophthalmoplegia. Usually, treatment with high-dose corticosteroids has been found to resolve the eye derangement in INO (9). This treatment was also found to be effective with our patient.
Three types of pathological changes have been reported in SLE-related myelopathy (2). The first is an extensive spinal cord softening limited to a specific cord level, accompanied by vascular alterations ranging from perivasculitis to thrombosis. The second is a large subdural hematoma in the spinal cord without vasculopathy. The third, the most common change, is a peripheral white matter degeneration, often at multiple spinal cord levels, which is called subpial leukomyelopathy. Pathogenic mechanisms of TM in SLE include vascular injuries secondary to immune-complex mediated vasculitis, non-vascular injuries due to antineuronal antibodies, and white matter degeneration (14). In addition, longitudinal myelitis might be related to either a vascular occlusive phenomena of the spinal cord associated with antiphospholipid antibody (aPL) or to a direct interaction between aPL and spinal cord phospholipids (15). Though we did not perform assays for antineuronal and antiribosomal P antibodies, it is quite likely that humoral factors other than aPL were implicated in the pathogenesis of the TM in this case because of negative results of aPL. Further research will be necessary to delineate the precise relationship between these laboratory parameters and spinal cord disease in TM.
The outcome of TM associated with SLE is favorable (16). It has been postulated that aggressive treatment early in the course of the disease is crucial for a favorable response (17). Therapy with i.v. pulsed cyclophosphamide, when given in conjunction with high dose corticosteroids, has been successful in TM (18). However, Tellez-Zenteno et al. (5) indicated that aggressive treatment with high dose corticosteroids and cyclophosphamide can not prove useful in the treatment of longitudinal myelitis, and found that the outcome is unfavorable in most cases. Also, Ropper and Poskanzer (3) emphasized that a hyperacute, catastrophic onset with the development of a total neurological deficit within 12 hr results in a poor outcome. Our patient received prompt treatment with corticosteroid and i.v. pulsed cyclophosphamide resulting in the restoration of a respiratory failure and the partial return of function of the upper extremities. However, neurological deficit below the T10 level, and bladder and colon paralysis persisted for six months.
In summary, this patient has the following characteristics. First, this is the first case of transverse myelitis involving the entire brain stem and spinal cord. The lesions are continuous from the midbrain to the conus medullaris longitudinally. Second, this patient's severe neurological symptoms had an abrupt catastrophic onset and rapidly progressed to respiratory failure within several days. Third, this patient is the first reported case of bilateral INO accompanied by transverse myelitis involving entire spinal cord longitudinally, and her MRI showed definite brainstem lesions deeply associated with INO.
 XML Download
XML Download