

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Sturge-Weber syndrome is a sporadic condition of mesodermal phakomatosis characterized by a portwine vascular nevus on the upper part of the face, leptomeningeal angiomatosis that involves one or both hemispheres, choroidal vascular lesions associated with glaucoma, seizures, neurologic deterioration, and eventual neurodevelopmental delay (1).
Klippel-Trenaunay-Weber syndrome, also a mesodermal phakomatosis, is defined by a triad of cutaneous and visceral hemangiomas, which can appear at any site of the body, venous varicosities, and soft tissue or bone hypertrophy (2). Overlap between Sturge-Weber syndrome and Klippel-Trenaunay-Weber syndrome is recognized (3). Klippel-Trenaunay-Weber syndrome is a nonhereditary condition that shares clinical features with Sturge-Weber syndrome and over 40 cases of combined Klippel-Trenaunay-Weber syndrome and Sturge-Weber syndrome have now been described (4).
Phakomatosis pigmentovascularis is a distinctive association of cutaneous hemangiomas and melanocytic nevi. The hemangiomas consist of extensive nevus flammeus, while the melanocytic lesions may be aberrant mongolian spots, nevus of Ota, and nevus of Ito. Nevus of Ota is a permanent, non-homogeneous bluish gray, black or brown lesion on the facial skin limited to the area innervated by the first and second divisions of the trigeminal nerve, with frequent association of the ipsilateral ocular pigmentation.
We experienced an unusual case of male infant with features of both Sturge-Weber syndrome and Klippel-Trenaunay-Weber syndrome, who presented with nevus flammeus on the face, back and leg, leptomeningeal angiomatosis on right hemisphere, hypertrophy of the right leg, hemiconvulsion, congenital glaucoma and nevus of Ota.
CASE REPORT
A 4-month-old male infant presented with nevus flammeus on the right leg and both part of the face and back, hypertrophy of the right leg and first attack of hemiconvulsion on the left. A diagnosis of Sturge-Weber syndrome was made during neonatal period on the basis of portwine vascular nevus on the upper part of the face, leptomeningeal angiomatosis that involves right hemisphere, choroidal vascular lesions associated with glaucoma. He had not taken any other form of medication in the previous 3 months.
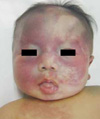
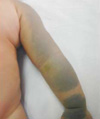
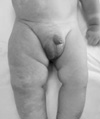
His physical examination showed nevus flammeus diffusely involving the both side of his face and extending to the both lower eyelids and cheeks. There were bilateral oculodermal melanosis and an area of bluish gray pigmentation of the episclera (Fig. 1). Diffuse bluish discoloration or spots were present on the left side of his shoulder, arm, and wrist (Fig. 2). His right lower limb showed a relative soft tissue hypertrophy and nevus flammeus (Fig. 3). There was no difference in length of both lower limb. But the circumference of his right thigh was 25 cm and that of his left thigh was 21.5 cm.
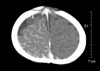
The hematologic, biochemical, and urinary laboratory tests and the chest and skull radiographs were normal. Abdominal ultrasonography showed no pathologic findings of the visceral organs. On cranial computed tomography scan, there was hyper-dense appearance secondary to meningeal venous angiomatous change on the right cerebral hemisphere (Fig. 4). The patient was diagnosed to have Sturge-Weber syndrome in association with Klippel-Trenaunay-Weber syndrome, according to the results of physical examination and radiologic evaluation.
Treatment with anticonvulsant was begun. But hemiseizures were recurred several times. His electroencephalography revealed low amplitude polyphasic spikes and waves activity, and voltage suppression in right hemisphere. The frequency of seizure was reduced probably due to anticonvulsants, excegran, acetazolamide and corticosteroid. At the time of discharge his medical regime included excegran and acetazoleamide.
DISCUSSION
Klippel-Trenaunay-Weber syndrome is present at birth and usually involves a lower extremity but may involve more than one and portions of trunk or face. Enlargement of the soft tissues may be gradual and may involve the entire extremity, a portion of it, or selected digits. Thick-walled venous varicosities typically become apparent ipsilateral to the vascular malformation after child begins to ambulate. Supportive care includes compression bandages for varicosities and surgical treatment may help selected patients. Associated complication include dislocation of joint, congestive heart failure, cerebral arteriovenous malformations, spinal arteriovenous malformations, macrocephaly, microcephaly, thombophlebitis, hematuria, rectal bleeding and orbitofrontal varices (5, 6).
Sturge-Weber syndrome is a rare, sporadic disease with a potentially progressive course. Facial and leptomeningeal angioma are usually, but not necessarily, on the same side (7). About 70% of patients with epilepsy have their first seizure within the first year of life. The initial seizures often involve the hemibody contralateral to the pial angioma and early seizures are triggered by fever in about one third of cases (8). Almost all the early seizures qualify as status epilepticus. The outcome after the first seizures is variable, but long term follow-up have shown that the seizure are often well controlled. Interictal EEG in Sturge-Weber syndrome shows focal and unilateral depression of the background activity over the area of leptomeningeal agiomatosis (9, 10). Focal epileptiform abnormalities are infrequent in infants, even in those with frequent seizures (10). Surgery should be considered early when medical treatment fails. The earlier the surgery with respect to seizure onset, the less the chance of increased neurologic deterioration (11).
Nevus of Ota is more common in females and in Asian and black patients, and differs from a Mongolian spots, not only by its distribution but also by having a speckled rather than a uniform appearance. Malignant change is rare and laser therapy may effectively decrease the pigmentation.
Four types of phakomatosis pigmentovascularis have been recognized, each of which are subgrouped further by the abscence (type a) or presence (type b) of systemic organ involvement. All types of phakomatosis pigmentovascularis have nevus flammeus as a component. In addition to nevus flammeus, type I has an epidermal nevus, type II has blue spots with or without nevus anemicus, type III has nevus spilus, with or without nevus anemicus, type IV has blue spots and nevus spilus with or without nevus anemicus. Our case can be classified as type IIa, according to the classification of phakomatosis pigmentovascularis. Phakomatosis pigmentovascularis is a rare disorder characterized by the association of a nevus flammeus and melanocytic lesions. Phakomatosis pigmentovascularis was first described as association of cutaneous hemangioma and melanotic or epidermal nevi by Ota in 1939 (12). Abnormalities in the neural regulations of blood vessels may be important in the development of the vascular component of phakomatosis pigmentovascularis. Anomaly in the migration of the neural crest-derived melanocytes would result in nevus of Ota, Mongolian spots and nevus spilus. There is double heterozygosity with the recessive vascular mutation on chromosome and the pigmentary mutation on the homologous chromosome. During embryogenesis, somatic recombination or crossing-over occurs between the homologous chromosomes, resulting in 2 different cell populations, each being homozygous for either allele. Associated clinical manifestation of the above-mentioned neurophacomatosis include scoliosis, renal angiomas, moyamoya disease, aplastic anemia, abdominal lymphangioma, chylothorax, deafness, chronic thyroiditis, pyogenic granuloma and mental retardation.
There is no specific curative treatment for this combined disorder. Recognition of possible underlying systemic and local anomalies and complications dictates management.
We present a 4-monthold baby boy who is considered as an interesting case of Sturge-Weber syndrome in association with Klippel-Trenaunay-Weber syndrome and phakomatosis pigmentovascularis. To the best of our knowledge, Klippel-Trenaunay-Weber syndrome has been reported many times, and the only one case of Sturge-Weber syndrome in association with Klippel-Trenaunay-Weber syndrome and phakomatosis pigmentovascularis has been reported in 13-yr-old girl (13). But the infantile case of the above-mentioned combined neurophacomatosis has not been reported. In infantile case there were no venous varicosities and bony hypertrophy of the affected extremities.
 XML Download
XML Download