

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Clinical manifestations of camptodactyly-arthropathy-coxa vara-pericarditis syndrome (CACP) include congenital or early-onset camptodactyly, noninflammatory arthropathy with synovial hyperplasia, progressive coxa vara deformity, and noninflammatory pericardial effusion. A CACP locus has been assigned to human chromosome region 1q25-31 by homozygosity mapping (1), and the syndrome is an autosomal recessive condition (2).
We report a case of CACP in a 10-yr-old boy with early-onset camptodactyly, noninflammatory arthropathy, coxa vara deformity, pericardial effusion, and without familial aggregation.
CASE REPORT
A 10-yr-old boy was admitted due to deformity of fingers and toes in August 2003. In July 1999, he was referred to our hospital because of joint swelling, pericardial effusion. Synovial fluid finding did not show inflammatory nature, and synovial biopsy revealed synovial hyperplasia without significant inflammation (Fig. 1). Ultrasound examination revealed hepatomegaly, but not splenomegaly. He had non-inflammatory arthropathy, pericardial effusion, and coxa vara. We decided to observe with conservative treatment.
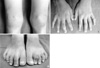
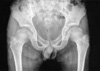
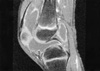
Months prior to admission, camptodactyly of fingers and toes was developed and gradually progressed, and he was referred to our hospital again. Physical examination showed a blood pressure 100/60 mmHg, pulse rate 90/min, temperature 36.0℃, and normal respiration pattern. Swelling of both knees, ankles, elbows and wrists and deformity of fingers and toes were observed (Fig. 2). There was no evidence of fever, lymphadenopathy and skin rash. His family members did not have musculoskeletal abnormality. Laboratory findings revealed a white blood cell 5,500/µL; hemoglobin 10.9 g/dL; platelet 194,000/µL; erythrocyte sedimentation rate 9 mm/hr, C-reactive protein 0.1 mg/dL (reference range 0.1-0.8). Tests for rheumatoid factor, antinuclear antibody and HLA-B27 were all negative. Liver function test, serum creatinine, urinalysis were in normal ranges. Synovial fluid analysis from knee joints showed a white blood cell 400/µL, red blood cell 2,450/µL, protein 2.5 g/dL, LDH 432 U/L. Chest radiography showed cardiomegaly, and pelvis AP revealed broad short femoral neck and coxa vara (Fig. 3). The plain radiography of both hands and both feet showed flexion deformity of fingers and toes (Fig. 4). Knee MRI showed large amount joint fluid and thin rim-like enhancement of the fluid filled bursae (Fig. 5). The echocardiogram showed moderate amount of pericardial effusion (Fig. 6). His chromosomal study was normal. He was diagnosed as having CACP and would be followed up whether camptodactyly and pericardial effusion would progress or not.
DISCUSSION
CACP is characterized by congenital or early-onset camptodactyly,
childhood-onset noninflammatory arthropathy associated
with synovial hyperplasia, progressive coxa vara deformity
and noninflammatory pericardial or pleural effusion.
The definition of camptodactyly is a congenital or acquired
nontraumatic flexion deformity of the proximal interphalangeal
(PIP) joint of one or several fingers (3). Camptodactyly
in CACP is usually bilateral and congenital, but in some
cases, it develops in early childhood. The degree of contracture
need not be equal in both and the deformity may progress
or not improve 3). Camptodactyly may be present as
an isolated entity or part of a spectrum of congenital anomalies.
Camptodactyly may be present in congenital anomalies
such as trisomy 13, oculo-dental-digital, oro-facial-digital,
cerebro-hepato-renal, Catel Manzke, Pena-Shokeir I syndromes
(3-5), and must be differentiated from a boutonniere deformity,
Dupuytren's contracture, a trigger finger, congenital
absence of the extensor mechanism (3). Nonoperative therapy
is effective in managing camptodactyly. Splinting is a valuable
tool in the initial management of the camptodactyly.
Tenolysis and tenosynovectomy is beneficial in some patients
(6, 7).
Arthropathy principally involves large joint such as elbows,
hips, knees, and ankles. Synovial fluid analysis reveals non-inflammatory
findings. Histopathologic analysis of synovial
tissue reveals pronounced hyperplasia of synovium without
evidence of inflammatory cell infiltration or vasculitis, while
synovial hyperplasia in rheumatoid arthritis is associated with
chronic inflammation. MRI finding of involved joints showed
only prominence of cartilage with normal menisci and cruciate
ligaments in one study (8), and rim-like enhancement
of the fluid filled bursae at T1 weighted image before and
after contrast enhancement (9). Enhancement is related to
the presence of inflammatory tissue, but the presence of rimlike
enhancement means the noninflammatory features of
CACP. The rim-like enhancement can distinguish between
CACP and juvenile rheumatoid arthritis (JRA) based on the
homogenous or multinucleated enhancement in JRA (10,
11). Therefore, MRI is regarded as a useful diagnostic tool
which differentiates the CACP from other childhood connective
tissue disease such as JRA (9).
The presence of coxa vara is noted in 50% of published
CACP cases (1), and in one study, 90% of cases have cova vara
deformity (9). The long-term follow-up of CACP patients
revealed the hip and spine involvement in some cases (12).
Noninflammatory pericarditis has been reported in up to
30% of CACP (1), and it may be mild and self-limited. But
it may be necessary to perform a pericardiocentesis or pericardiectomy
in life-threatening cases (2, 13-15).
CACP is a genetically homogenous condition despite clinical
variability and differences in ethnic and geographic origins
(12), and it has autosomal recessive mode of inheritance
(2). A CACP locus is assigned to a 1.9-cM interval on human
chromosome 1q25-31 by homozygosity mapping (1). Marcelino
et al. identified mutations in a gene (CACP) encoding a
secreted proteoglycan as the cause of CACP (16).
Although some cases of CACP were reported in Caucasian,
Egyptian, Saudi Arabian, but there has been no report in
Korea. The reason why CACP is rare in Korea may be due
to rarity of consanguineous marriage, which increases the incidence
of autosomal recessive disease. In this case, sporadic gene
mutation might be responsible for the disease, because his
family has no arthropathy or joint deformity. Whenever we
see juvenile patients with noninflammatory arthropathy, congenital
musculoskeletal disease such as CACP should be considered.



 XML Download
XML Download