

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Aldosterone is a steroid hormone released by the adrenal cortex. Classically, it acts on kidney, colon and sweat/salivary glands to promote unidirectional sodium transport (1). Recent studies, however, suggested that aldosterone might have its direct effect on the heart. In this regard, aldosterone was reported to alter cardiac remodeling and promote fibrosis in vivo (2, 3). Also, it was shown that aldosterone promotes collagen production in cardiac fibroblasts in vitro (4-7). In an animal model of hyperaldosteronism, significant cardiac fibrosis was observed (8). Consistent with these findings, convincing evidence for a local cardiac aldosterone system was reported in the normal rat heart (9). Moreover, recent clinical trial (the Randomized Aldactone Evaluation Study [RALES]) demonstrated that a mineralocorticoid receptor antagonist, spironolactone, has a significant beneficial effect on mortality and morbidity of patients with heart failure (10), suggesting a crucial role for aldosterone.
Connective tissue growth factor (CTGF) is a cysteine-rich, 38-kDa polypeptide that is induced by dexamethasone, transforming growth factor (TGF)-β1, vascular endothelial growth factor (VEGF), and thrombin. It was originally isolated from human umbilical vein endothelial cells (11) and was subsequently found in many cells including fibroblasts, smooth muscle cells, and chondrocytes (12-14). CTGF is involved in many cellular processes underlying fibrosis such as cell proliferation, migration, adhesion, and the synthesis of extracellular matrix (ECM) (15). Because CTGF induces fibronectin and collagen type I, which are the molecules abnormally deposited in fibrotic lesions of major organs such as liver, kidney, lung, and skin (16-19), it was suggested to be involved in the development of fibrotic pathology (20).
With regard to the role of CTGF in the heart, in situ hybridization studies showed that CTGF mRNA is increased in cardiac myocytes and mesenchymal cells in the infarct zone of rat hearts following myocardial infarction (21). Significant upregulation of CTGF was also detected in the heart of patients diagnosed with ischemic heart disease (22). In addition, it was demonstrated that CTGF expression is induced by TGF-β in cardiac fibroblasts and cardiac myocytes where concomitant increase in the production of fibronectin, collagen, and plasminogen activator inhibitor-1 was observed (22).
Despite these known behaviors of CTGF and aldosterone, studies on the direct relationship between the two molecules have been limited. Here, we examined the effect of aldosterone on CTGF expression in rat embryonic ventricular myocytes (H9c2). The underlying mechanism for aldosterone-mediated CTGF upregulation was also studied to have further insight into the CTGF induction pathway. We found that CTGF expression is increased by aldosterone in a dose- and time-dependent manner and that both mineralocorticoid receptor and p38 MAPK pathway are involved in this upregulation. Moreover, we present evidence that mineralocorticoid receptor is connected to p38 MAPK pathway. Our findings warrant further study on the signaling cascades that lead to CTGF induction by aldosterone.
MATERIALS AND METHODS
Cell culture
Rat embryonic ventricular myocytes (H9c2) were cultured in a humidified atmosphere (5% CO2) at 37℃ in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (GIBCO; Grand Island, NY, U.S.A.) supplemented with penicillin (100 units/mL) and streptomycin (10 µg/mL). The cells were seeded in 60 mm plate and cultured for 24 hr in a serum-free DMEM until the addition of aldosterone. Inhibitors were added to the cell 30 min before aldosterone treatment.
Northern blot analysis
Total cellular RNA was isolated using TRIZOL reagent (GIBCO). The integrity of RNA was checked on 1% agarose/formaldehyde gel. The RNA was quantified by spectrophotometer at 260 nm. For Northern blot analysis, 10 µg of the RNA was run in the gel followed by transfer to nitrocellulose membrane (Schleicher & Schuell; Dassel, Germany) and fixation using an UV cross-linker (Stratagene; La Jolla, CA, U.S.A.) at 120,000 µJ/cm2. Prehybridization and hybridization were performed at 42℃ in 5×SSPE (1×SSPE=0.18 M NaCl, 10 mM NaH2PO4, 1 mM EDTA, pH 8.0) solution containing 50% formamide, 10×Denhardt's solution, and 0.5% (w/v) SDS. After prehybridization for 2 hr, the membranes were hybridized with the 32P-labeled CTGF cDNA or 18S rRNA probes. The latter was used to normalize RNA loading. The membranes were washed with low stringency buffer, 2×SSC solution (1×SSC=150 mM NaCl, 15 mM C6H5Na3O7·2H2O) containing 0.1% (w/v) SDS, for 30 min at room temperature followed by washing with high stringency buffer (0.5×SSC containing 0.1% (w/v) SDS) for 30 min at 50℃. They were then washed with 0.2×SSC for 2 hr at 55℃. The films were exposed at -80℃ and the intensities of the CTGF and 18S rRNA bands were quantified using densitometer.
Western blot analysis
The cells were harvested in lysis buffer A (50 mM Tris-HCl, pH 7.4, 1% (v/v) NP-40, 0.25% (w/v) sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 µg/mL aprotinin, 1 µg/mL leupeptin, and 1 µg/mL pepstatin). Then the lysates were incubated on ice for 30 min with occasional mixing followed by centrifugation at 13,000 g for 10 min at 4℃. Protein concentrations were determined by the commercial kit (Bio-Rad; Hercules, CA, U.S.A.). After boiling for 5 min in SDS sample buffer, the lysates (20 µg) were subjected to 10% SDS-polyacrylamide gel electrophoresis and transferred to Hybond-P membrane (Amersham) using electro-transfer apparatus (Bio-Rad). The membranes were incubated for 1 hr in 0.1% (v/v) Tween 20-Tris buffered saline (TBST) solution containing 5% (w/v) skim milk for blocking. They were then incubated with primary antibody (polyclonal rabbit antibodies for the phosphorylated and total forms of ERK1/2, p38 MAPK, and JNK from Cell Signaling Technologies, Beverly, MA, U.S.A.) at 1:1,000 dilution in blocking solution at 4℃ overnight. After washing three times in TBST for 10 min, membranes were incubated with 1:2,500 dilution of goat anti-rabbit secondary antibody conjugated to horseradish peroxidase (Amersham, Piscataway, NJ, U.S.A.) for 1 hr at room temperature. The blots were washed three times in TBST and developed using chemiluminescence kit (ECL; Amersham).
RESULTS
CTGF is upregulated by aldosterone
Despite many reports on the role of aldosterone and CTGF in tissue fibrosis (3, 4, 16-19, 23), the direct relationship between the two molecules has remained unknown. To elucidate the effect of aldosterone on CTGF expression, we first investigated whether aldosterone can increase CTGF expression in rat embryonic ventricular myocytes, H9c2. When the cells were treated with various (1 nM to 10 µM) concentrations of aldosterone (Sigma, St. Louis, MO, U.S.A.) for 2 hr, CTGF mRNA was significantly upregulated above basal level by aldosterone at concentrations of 10 nM or higher (Fig. 1A). The amounts of 18S rRNA were similar among different samples. Next, we examined the time course of CTGF upregulation by aldosterone (Fig. 1B). The peak expression of CTGF was seen at 2 hr after aldosterone treatment and further incubation did not help to increase the level of CTGF mRNA. These data indicate that CTGF mRNA is upregulated by aldosterone in a dose- and time-dependent manner.
Mineralocorticoid receptor is involved in CTGF upregulation
To explore molecular mechanism of the aldosterone-induced CTGF upregulation, we first examined the role of mineralocorticoid receptor by using spironolactone, a mineralocorticoid receptor antagonist. Spironolactone can prevent myocardial fibrosis in a rat model of primary and secondary hypertension even without blood pressure effects (24, 25) and thus would be expected to prevent upregulation of CTGF. The H9c2 cells were treated with different (0.1 µM to 2.5 µM) concentrations of spironolactone (Sigma) for 30 min and then exposed to 0.5 µM aldosterone. As shown in Fig. 2, pre-treatment of spironolactone at 5-fold excess of aldosterone blocked the transcriptional upregulation of CTGF by aldosterone. This result suggests that mineralocorticoid receptor is involved in CTGF induction by aldosterone.
Aldosterone activates ERK, JNK, and p38 MAPK
MAPKs such as extracellular signal-regulated kinases (ERK), c-Jun NH2-terminal kinases (JNK), and p38 MAPK play a pivotal role in transducing extracellular signal (26). To investigate involvement of MAPK in aldosterone-mediated CTGF induction, we examined activation kinetics of each MAPK in H9c2 cells by Western blot analysis using phospho-specific antibodies (Fig. 3). Compared with the control cells at 0 min, aldosterone treatment resulted in strong activation of ERK1/2, reaching plateau at 20 min. The more transient pattern of activation was observed in p38 MAPK with maximum at 10 min after aldosterone treatment. The JNK was also activated by aldosterone with peak at 60 min. These results indicate that aldosterone activates all three MAPK pathways.
p38 MAPK pathway is involved in the CTGF induction by aldosterone
We next investigated involvement of each MAPK pathway in CTGF upregulation by aldosterone. To this end, inhibitors of ERK1/2, p38 MAPK, and JNK pathway (PD098059 (27), SB203580 (28), and JNK inhibitor I (29), respectively; Cal-Biochem, La Jolla, CA) were used. After pre-incubation in the presence of the inhibitor for 30 min, the cells were exposed to aldosterone (1 µM). As shown in Fig. 4, aldosterone-mediated CTGF induction was significantly blocked by SB203580, but not by PD098059 and JNK inhibitor I. This result indicates that p38 MAPK cascade as well as other pathways inhibited by SB203580 may be important for the CTGF upregulation following aldosterone treatment. On the other hand, JNK inhibitor I and PD098059 decreased the basal level of CTGF expression (lane 7 and lane 5, Fig. 4) with no loss in induction fold, suggesting that ERK1/2 and JNK pathways may be required for basal expression of CTGF.
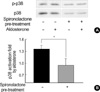
Effect of spironolactone on p38 MAPK activation
Because aldosterone upregulated CTGF expression via both mineralocorticoid receptor and p38 MAPK pathway, we examined the effect of spironolactone treatment on p38 MAPK activation (Fig. 5). Interestingly, pre-treatment of spironolactone at 5-fold excess of aldosterone completely blocked aldosterone-induced activation of p38 MAPK, indicating that mineralocorticoid receptor mechanism is linked to MAPK pathway.
DISCUSSION
Despite years of studies on aldosterone and CTGF with respect to fibrosis, the direct relationship between the two molecules has remained unelucidated. In the present study, we investigated the effect of aldosterone on CTGF in rat embryonic ventricular myocytes and found that CTGF expression is increased by aldosterone in a time- and dose-dependent manner. Our observation that aldosterone upregulates CTGF expression in heart-derived myocytes is supported in part by the recent microarray study of another group who added CTGF to the list of early aldosterone-responsive genes in mouse inner medullary collecting duct cells (30). We also found that both mineralocorticoid receptor and p38 MAPK pathway are important for aldosterone-mediated CTGF upregulation. Lastly, we present circumstantial evidence that there exists cross-talk between mineralocorticoid receptor cascade and p38 MAPK activation.
Aldosterone and other adrenal corticosteroids exert many of their physiological actions through modulation of gene expression. The classic genomic action of aldosterone is characterized by a latency of onset and by the sensitivity to specific antagonist of intracellular receptor such as spironolactone. Our results here strongly suggest that the classic genomic action of aldosterone was involved in the upregulation of CTGF as the time course of CTGF upregulation indicated a substantial latency (Fig. 1B) and because pre-treatment of spironolactone blocked the induction of CTGF (Fig. 2). Moreover, homology searches of the CTGF gene for the transcription factor binding elements have identified at least three putative steroid-responsive elements (SREs) in the 5'UTR region of the CTGF which spans 1 kb from the transcriptional start site (data not shown). Since the genomic action of aldosterone involves interaction between the intracellular receptor-steroid complex and SRE located within promoter of target genes, these SREs might have acted as cis-enhancer elements in the upregulation of CTGF by aldosterone.
We also focused our attention on MAPKs as they are common participants in signal transduction pathways initiated by growth factors, cytokines, stresses, and various pharmacological compounds (31, 32). We demonstrate here that p38 MAPK pathway is important for the aldosterone-mediated CTGF induction and that ERK1/2 and JNK pathways may be required for basal expression of CTGF in ventricular myocytes (Fig. 4). Consistent with our data, it was previously reported that p38 MAPK inhibitor, FR-167653, could significantly suppress the expression of CTGF mRNA during the later phase of bleomycin-induced pulmonary fibrosis and thus ameliorates the fibrosis in a murine model (33). On the other hand, it was recently reported that aldosterone augments JNK activation induced by endothelin-1 in cardiac myocytes (34). Also, it was shown that aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-RasA and ERK1/2 signaling, which are thought to be involved in the pathological actions of aldosterone on the heart (7). These different findings suggest the presence of complicated signaling processes and the interactions involved in the pathogenesis of cardiac fibrosis.
CTGF expression is also induced by TGF-β in cardiac fibroblasts and cardiac myocytes (22). Based on our observation that aldosterone upregulates CTGF, it can be presumed that the role of TGF-β and aldosterone would overlap as the inducer of cardiac fibrosis. Moreover, the same induction mechanisms including p38 MAPK pathway might be involved in CTGF induction by TGF-β. Although aldosterone might exert its indirect effect on TGF-β via CTGF, it would be interesting to see in detail the interplay between TGF-β and aldosterone in the progression of cardiac fibrosis.
With regard to the effect of spironolactone on MAPK activation, the spironolactone pre-treatment resulted in blockade of p38 MAPK activation (Fig. 5). One simple explanation for this observation would be that mineralocorticoid receptor acts at upstream of p38 MAPK pathway, thus relaying the signal to p38 MAPK upon binding of aldosterone. Another possibility is that spironolactone might directly inhibit activation of p38 MAPK via yet unknown mechanisms. When a group of responses that are mediated by receptors of aldosterone and several different pathways are taken into account, definitive explanation of the above observation may be difficult at present and will have to wait for the more detailed studies on the cross-talk between divergent signaling cascades.
One of the limitations of the present study is that the H9c2 embryonic ventricular myocytes used in this study cannot actually represent the adult cardiac myocytes. Because the H9c2 cells can differ from adult cardiomyocytes by a number of features, the conclusions made from the experiments in H9c2 can hardly be extended to adult cells. Another caveat is that cardiac myocytes synthesize only collagen IV, a minor membrane-bound form, and therefore do not contribute significantly to adult cardiac fibrosis via collagen production. Moreover, a direct induction of collagen synthesis has never been observed in other cardiac fibroblast or myocytes, greatly weakening the significance of these cells in cardiac fibrosis. Further studies on additional fibrosis factors such as matrix metalloproteinases and fibronectin as well as the interactions of several cell types involved are required to evaluate the overall importance of CTGF induction by aldosterone in cardiac fibrosis.
In conclusion, our study shows for the first time that the CTGF induction by aldosterone involves p38 MAPK cascade and mineralocorticoid receptor and that the two pathways are linked. Our findings set the stage for detailed studies on the signaling cascades that are initiated by aldosterone and lead to CTGF upregulation.




 XML Download
XML Download