

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Increases in apoptotic chondrocytes have been reported in rheumatoid arthritis (RA) and osteoarthritis (OA) cartilages in situ (1, 2), and since any conditions that compromise chondrocyte survival will have detrimental effects on the maintenance of proper articular cartilage, the mechanism of chondrocyte apoptosis has been the focus of interest recently as one of the pathogenetic factors leading to joint cartilage degradation. The signaling cascade that triggers apoptosis in chondrocytes has been extensively studied. Cultured chondrocytes undergo apoptosis in response to various stimuli, including serum deprivation (3), FAS-ligand or anti-FAS/CD95-antibodies (4), and nitric oxide (NO) donor sodium nitroprusside (5).
Among the cytokines that mediate cartilage degradation, tumor necrosis factor alpha (TNF-α) is unique in that it is related with the signaling mechanism of both matrix degradation and cell death. Signaling cascade of TNF-α cytotoxicity begins with the binding of TNF-α to the p55 TNF receptor (TNFR), receptor trimerization, the recruitment of TNFR-associated death protein and TNFR associated factor 2, Fasassociated protein with death domain (FADD), and caspase-8 (6). Both TNF-R55 and TNF-R75 are expressed constitutively on human articular chondrocytes (7). Human articular chondrocytes are resistant to TNF-alpha induced cell death and undergo cell death only when they are treated with TNF-alpha in the presence of transcription inhibitor or proteasome inhibitor (8). TNF-alpha receptor signaling activates numerous downstream signaling cascades which include caspase 8, NF-κB, and mitogen activated protein (MAP) kinases. Although the proapoptotic role of caspase 8 and anti-apoptotic role of NF-κB have been well delineated, the role of MAP kinase in TNF-alpha signaling in light of apoptosis induction is not settled. There are 3 distinct subtypes of MAP kinases identified in mammalian cells: extracellular signal-regulated kinases (ERK), c-Jun N-terminal protein kinase (JNK), and p38 kinases. A recent report showed that the inhibition of ERK-1/2 enhances sodium nitroprusside (SNP)-induced apoptosis, whereas the inhibition of p38 kinase blocks it in rabbit articular chondrocytes (9). The role of JNK activation in TNF-alpha mediated chondrocyte death signaling is not known.
The objective of this study was to delineate the pattern of JNK activation in TNF-alpha mediated chondrocyte death. We observed the influence of inhibition of JNK dephosphorylation, which leads to sustained activation of JNK, on TNF-alpha mediated chondrocyte death, as well as on the expression of apoptosis related proteins.
MATERIALS AND METHODS
Reagents
Recombinant human TNF-α was purchased from R&D (Minneapolis, MN, U.S.A.). Pan-caspase inhibitor (z-VAD-FMK) was purchased from Biomol (Plymouth Meeting, PA, U.S.A.). Anti-JNK, anti-IκB, and anti-phosphoIκB were purchased from New England Biolab (Beverly, MA, U.S.A.), anti-Bcl-2 from Transduction Laboratory (Lexington KY, U.S.A.), anti-Bax, anti-FLIP (Flice inhibitory protein) and anti-caspase-3 from Pharmingen (San Diego, CA, U.S.A.), and anti-p53, anti-p21, anti-MDM2, anti-p65 from Santa Cruz (Santa Cruz, CA, U.S.A.). All other reagents were obtained from Sigma (St. Louis, MO, U.S.A.) unless specified otherwise.
Chondrocyte culture
Cartilage samples were obtained from the tibial plateau of knee OA patients. Pieces of articular cartilage were cut, minced, and incubated with trypsin/collagenase (Sigma, St. Louis, MO) in DMEM until the fragments were digested. Released cells were seeded at 2×106/plate in 10 cm culture plates in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 1% L-glutamine, 1% Fungizone (Gibco, Grand Island, NY, U.S.A.) and penicillin/streptomycin (150 units/mL and 50 mg/mL each). After about 7-10 days, confluent chondrocytes were split once, seeded at high density, and these first passage chondrocytes were used in the subsequent experiments.
Induction of apoptosis
Before adding TNF-α, the medium containing 10% FCS was replaced with medium containing 0.5% FCS, and the chondrocytes were incubated overnight. TNF-α was added to the medium in a concentration of 10 ng/mL. The potentiation of apoptosis was attempted with the following compounds: actinomycin D, sodium vanadate, or Ro318220 by adding each compound to the media 1 hr prior to TNF-α.
SAPK/JNK assay
SAPK/JNK activity was measured using a solid phase kinase assay method. Briefly, 100 microgram of cell lysate was incubated with 2 microgram of GST-c-Jun beads (Cell signaling technology, Beverly, MA, U.S.A.) at 4℃ overnight with gentle rocking. The beads were then pelleted, washed and resuspended in kinase buffer (25 mM Tris pH 7.5, 5 mM β-glycerolphosphate, 2 mM DTT, 0.1 mM Na3VO4, 10 mM MgCl2) containing final concentrations of 100 µM ATP. After incubation at room temperature for 30 min, the reaction was terminated by adding SDS sample buffer. The proteins were separated on 12% SDS-PAGE, and blotted with phospho-c-Jun antibody (Cell signaling technology, Beverly, MA).
Quantification and verification of cell death
The amount of cell death was quantitated by using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5 diphenyltetrazdium bromide (MTT) assay, as described (10). Briefly, chondrocytes were seeded at 5×104/100 µL/well in 96 well microtiter plates, and incubated with TNF-α with or without various potentiators. MTT was added to each well to a final concentration of 0.125 mg/mL, and the plate incubated at 37℃ for 3 hr, after which, the formazan product was solubilized with 100 µL of dimethylsulfoxide (DMSO) and the optical density was read at a wavelength of 595 nm. The percentage of cell survival was calculated by taking the optical density of cells given a particular treatment, dividing this by the optical density of untreated, control cells, and then multiplying by 100. Apoptosis was also verified by flow cytometry. For analysis of subdiploid cells, chondrocytes were trypsinized after treatment and sedimented. Obtained cell pellets were fixed in 70% ethanol, and stained in 100 µg/mL propidium iodide solution containing 50 µg/mL RNase for 15 min. For annexin V-FITC/propidium iodide (PI) labeling, cells were collected and resuspended in cold binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) to a final concentration of 1×106/mL. Aliquots of 1×105 cells were incubated with 5 µL of annexin-V-FITC and 5 µg/mL of PI. After 15 min at room temperature, 400 µL of binding buffer was added before flow cytometric analysis. For each sample, 104 cells were analyzed on a FACS II flow cytometer (Becton Dickinson, Mountain View, CA, U.S.A.).
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts from chondrocytes were prepared from 2×106 cells as described previously with minor modifications (11). Protein content was measured and 5 µg portions of extracts were used for the binding reaction. A consensus double-stranded NF-κB probe was obtained from Promega (Madison, WI, U.S.A.), and end-labeled by using γ-32P-adenosine-5-triphosphate. Nuclear extracts were incubated in gel binding buffer (Promega, Madison, WI, U.S.A.) in a volume of 9 µL. Afterwards, the end-labeled probe was added (100,000 cpm/sample). Samples were then incubated for 20 min and loaded onto 4% nondenaturing polyacrylamide gel. Electrophoresis was run for 3 hr in a cold room. Protein complexes were identified by autoradiography.
Western blot
Cellular protein was extracted in lysis buffer containing 50 mM sodium acetate, pH 5.8, 10% v/v SDS, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and 1 µg/mL aprotinin at 4℃. Samples were electrophoresed on 12% SDS-PAGE gel, transfered, and Ponceau S staining of PVDF membrane was routinely performed to roughly confirm equal loading of protein before every Western blot procedure. Blots were blocked with Tris buffered saline (TBS) containing 5% nonfat milk at room temperature for 1 hr, and incubated with respective antibodies overnight at 4℃. The blots were then incubated with 1:5,000 peroxidase-conjugated goat anti[-mouse or anti-rabbit IgG (Biorad, Hercules, CA, U.S.A.) for 1 hr. Bound immunoglobulin was detected by using an enhanced chemiluminescence kit (Amersham, Bucks, U.K.).
RESULTS
The pattern of activation of the JNK in response to TNF-alpha
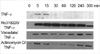
Vanadate and Ro318220 are protein phosphatase inhibitors, which is known to inhibit the tyrosine phosphatases (12). Treatment with vanadate and Ro318220 inactivate protein phosphatases that dephosphorylate JNK, thereby prolonging the activation of JNK by TNF-alpha (13). First, we tried to define the pattern of activation of the JNK in response to TNF-alpha in Ro318220 or vanadate pretreated chondrocytes. Treatment with 10 ng/mL of TNF-alpha led to prompt phosphorylation of JNK, first apparent after 5 min, and lasting about 15 min (Fig. 1). Activation of JNK, as detected by a solid phase kinase assay of phosphorylated c-Jun, was significantly prolonged in Ro318220 or vanadate pretreated chondrocytes, as was expected, exhibiting about 5 hr of phosphorylation. Actinomycin D, another potentiator of TNF-alpha mediated chondrocyte death (8) also led to prolonged activation of JNK. In this case, the first JNK phosphorylation band disappeared and then reappeared after 240 min of TNF-alpha treatment, lasting until 5 hr.
The influence of phosphatase inhibitors on TNF-alpha induced chondrocyte death
In order to define the influence of phosphatase inhibitors on chondrocyte death, we analyzed cell death in each treated chondrocytes. As was reported previously in human OA chonrocytes (8), the treatment of chondrocytes with TNF-alpha alone failed to reveal any significant degree of cell death. However, pretreatment with 500 µM of vanadate or 10 µM Ro318220 led to significant amount of cell death in chondrocytes treated with 10 ng/mL of TNF-alpha. To assess the extent of cell death according to incubation time, cell viability was determined by MTT assay after 12, 24, 36, and 48 hr of incubation (Fig. 2A). Pre-treatment with vanadate or Ro318220 significantly affected cell viability in a time dependent manner and cell death significantly increased 12 hr after treatment (p<0.0001). The extent of cell death was more profound in Ro318220 pretreated chondrocytes compared to vanadate pretreated chondrocytes. Treatment with Ro318220 or vanadate alone also decreased chondrocyte viability, but the extent of cell death was not as marked as in co-treatment with TNF-alpha. Apoptosis was also verified by flow cytometry analysis of subdiploid fraction (Fig. 2B) and annexin V/propidium iodide stating (Fig. 2C), but the percentage of apoptotic chondrocytes as revealed by this method tended to be lower compared to that analyzed with MTT assay. Caspase 3 processing was observed by immunoblot as early as 6 hr after Ro318220/TNF-α or vanadate/TNF-α treatment, which occurred before apoptotic cell death was discernible (Fig. 2D). In order to verify the involvement of the caspase cascade in Ro318220 or vanadate facilitated TNF-α mediated chondrocyte death, we performed an inhibition assay using a pan-caspase inhibitor, zVAD-FMK. As is shown in Fig. 2E, cell death by both Ro318220/TNF-α or vanadate/TNF-alpha was effectively inhibited by zVAD-FMK.
The influence of phosphatase inhibitors on TNF-alpha induced NF-κB activation
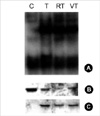
Next, we examined the influence of Ro318220 and vanadate on TNF-alpha induced NF-κB activation and IκB degradation in chondrocytes, which is related with survival signaling pathway in response to TNF-alpha. Chondrocytes responded by translocation of NF-κB and degradation of IκB after treatment with TNF-alpha, and pretreatment with Ro318220 or vanadate did not exhibit any significant influence on this response (Fig. 3) Phosphorylation of IκB was not affected, either.
The influence of phosphatase inhibitors on apoptosis-related molecules
Lastly, we investigated the influence of Ro318220 and vanadate on the level of apoptosis-related molecules. Cells were incubated for 2, 4, 6, 8, 12, and 18 hr in TNF-alpha with or without pretreatment and proteins were extracted. First, we investigated the levels of Bcl-2 family proteins. The levels of Mcl-1 and Bcl-2 began to decrease from 2 to 8 hr of culture in Ro318220 and vanadate pretreated chondrocytes while treatment with TNF-alpha alone did not exert any significant effect on these proteins (Fig. 4). The expression patterns of p53, MDM2, p21, FLICE inhibitory protein (FLIP), Bax, Bcl-XL, and XIAP was not affected in any of the treatment condition (data not shown).
DISCUSSION
The objective of this study was to delineate the correlation of JNK activation with TNF-alpha mediated chondrocyte death by using chemical inhibitors of JNK dephosphorylation. Our study shows that TNF-alpha signaling leads to chondrocyte death in the presence of phosphatase inhibitors, which induces prolonged JNK activation, and this pathway is dependent on caspase cascade.
Activation of NF-κB by TNF-alpha receptor associated factor 2 (TRAF2) is postulated to serve as a primary mechanism to protect cells against apoptosis (14). TNF-alpha also plays a role of protection in apoptosis induced by other stimuli. A recent report (15), showed that TNF-alpha protects chondrocytes from death induced by NO, and this protection was conferred by 3 mediators, NF-κB, phosphatydylinositol-3 kinase (PI-3K) which possibly could also stimulate NF-κB transcription activity, and cyclooxygenase-2. MAP kinases lies in proximity to NF-κB in TNF-α signaling, and among the 3 subtypes identified in mammalian cells, JNK and p38 kinases are strongly activated by extra- or intracellular stress and inflammatory cytokines (16). It is thought that activation of JNK and p38 kinases generally promotes an inhibition of cell growth or promotion of cell death, while ERK is usually strongly activated by growth factors and hormones that stimulate cell growth and is involved in the regulation of cell proliferation (17). Strong and prolonged activation of JNK has been reported in response to a variety of stresses including UV light, ionizing radiation and hydrogen peroxide, all of which can induce apoptosis (18, 19). Because TNF-alpha strongly activates JNK while it activates ERK only weakly in many cells, it was postulated that JNK activation is involved in TNF-alpha induced apoptosis (20). The role of JNK pathway in TNF-alpha mediated signaling in chondrocytes has not been elucidated, however. We have employed chemical inhibitors of protein phosphatases, Ro318220 and vanadate in order to inhibit dephosphorylation of activated JNK potently, and our results show that inhibition of JNK dephosphorylation and resulting prolongation of JNK activation is associated with chondrocyte death. In addition, actinomycin D, another potentiator of TNF-mediated chondrocyte death also led to significant prolongation of JNK activation. An obligatory role of the JNK pathway has been documented in various stress-induced apoptosis models. The duration of JNK activation is thought to be a critical factor determining cell survival or apoptosis, with transient activation leading to cell proliferation and prolonged activation leading to apoptosis (18). The pattern of JNK activation induced by actinomycin D is reminiscent of JNK activation induced by Fas in a neuroblastoma cell line, which was biphasic with first peak at 15 min and second peak starting 2 hr after treatment (21). From this result, it can be hypothesized that actinomycin D also blocks the expression of a phosphatase that limits the activation of JNK. The involvement of JNK in TNF-alpha mediated apoptosis is not without debate (22, 23), however. A recent study has shown that JNK is not required for TNF-alpha mediated cell death and mouse embryonic fibroblast deficient for JNK1 and JNK2 are somewhat more sensitive to TNF-alpha, suggesting that JNK may play a role as survival factor (24). It is possible that low or basal levels of JNK provide protection against cell death while sustained activation of JNK overrides the anti-apoptotic signals to promote cell death. In addition, different means of JNK activation and/or cell context may lead to different responses. Despite the fact that sustained activation of JNK promotes cell death, the molecular basis of how JNK contributes to TNF-alpha mediated apoptosis remains to be elucidated.
In this study, the amount of cell death detected by MTT assay tended to be higher than that by flow cytometry. The extent of cell death can vary according to the method of analysis, and in our previous report, we found out that the proportion of dead cell is higher by MTT assay compared to flow cytometry (25). This discrepancy might stem from the loss of dead cells in the process of cell collection inherent in flow cytometry methodology. It is also possible that some of the cell death detected by MTT assay represents non-apoptotic cell death.
Recently, we have observed that chondocytes transduced with adenovirus encoding IkB-superreprresor, which exhibits resistance to degradation resulting in selective inhibition of NF-κB activation, are induced to undergo apoptosis in response to TNF-alpha. JNK activation is prolonged in this condition of chondrocyte death (manuscript submitted). The interrelationship between JNK and NF-κB was suggested by another report showing that the IKK/NF-kappaB pathway negatively modulates TNF-alpha-mediated JNK activation, partly through NF-kappaB-induced XIAP, which is specific to TNF-alpha signaling, and which contributes to inhibition of apoptosis (26). In order to verify whether inhibition of JNK inactivation might affect NF-κB activation, conversely, we looked at the pattern of NF-κB activation and IκB degradation. Pretreatment with phosphatase inhibitors did not have any discernible effects on NF-κB activation.
Among the apoptosis related proteins, the expression of Bcl-2 family proteins was most markedly changed. These included the proteins related with protection of apoptosis, such as Mcl-1 and Bcl-2. The involvement of the Bcl-2 family proteins in protection from apoptosis induced by JNK activation was previously reported (27, 28).
We used chemical inhibitors of phosphatase and it could be argued that the effect of JNK activation apart from the primary stress induced damage or from other stress-induced signaling pathway cannot be separated. In order to serve this goal, a constitutively active JNK mutant which allows selective activation of the JNK cascade in the absence of any cellular stress should be employed. In a recent report employing such a mutant, deltaMEKK1:ER, it was revealed that additional inhibition of the PI3K cell survival pathway is necessary to induce apoptosis in CC139 cells, even in conditions of sustained activation of JNK (27).
In summary, we observed that sustained activation of JNK induced by phosphatase inhibitors or actinomycin D after TNF-α treatment is associated with chondrocyte death. This effect may be due to blocking the expression of a TNF-α induced phosphatase that inactivates JNK. The expression of protecting molecules of apoptosis, Mcl-1 and Bcl-2 was downregulated.


 XML Download
XML Download