

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Severe Epstein-Barr virus (EBV) disease has been observed increasingly in immune deficient hosts having congenital immunodeficiency syndrome or acquired immune suppression. In an immune competent host, EBV may be eliminated by cytotoxic T cells evoked by the EBV antigen, or the virus can survive in the human body through a latent infection by maintaining a balance with the host. Sometimes, a fulminant EBV disease occurs in previously healthy hosts. Fulminant EBV diseases have been reported under various names including fatal EBV-associated hemophagocytic syndrome or severe chronic active EBV infection (1, 2). Severe chronic active EBV infection commonly occurs in children or adolescents, but rarely in adults, and is characterized by chronic illness lasting more than 6 months and an elevated titer of EBV-associated antibodies, pancytopenia, hemophagocytosis, disseminated intravascular coagulation, hepatosplenomegaly, and lymphadenopathy (3). Unlike classic infectious mononucleosis, T-lymphocytes or NK cells instead of the B-cell harbor the EBV genome, and some cases progress to T or NK-lymphoproliferative disease (4).
We report a case of chronic active EBV infection occurring in a 61-yr-old Korean woman with a reappraisal of the current diagnostic criteria.
CASE REPORT
The patient was referred to the Hemato-oncology Department because of a fever of unknown origin. She had been suffering from chronic hepatitis C since 4 yr prior to her visit, when elevated AST/ALT and positive HCV-RNA of peripheral blood were noted. IFN-α and ribavirin had been intermittently administered at an outside hospital. Two months prior to the transfer to our hospital, she had a fever that was not improved by discontinuation of IFN-α and ribavirin.
On admission, her body temperature was 38.5℃, her blood pressure 140/80 mmHg, and her respiration rate 20/min. She appeared chronically ill. Blood findings were Hb 7.9 g/dL, platelet 61,000/µL, and WBC 3,900/µL with differentials of segmented neutrophils 48.4%, lymphocytes 34.6%, and monocyte 14.9%. The reticulocyte count was 6.6% and ESR 33 mm/hr. A liver function test revealed total bilirubin of 1 mg/dL, AST 46 µL, ALT 19 µL, and γ-GT 36 µL. Total protein was 8.7 g/dL, globulin 6.3 g/dL, and albumin 2.4 g/dL. An immunoelectrophoresis of the blood protein revealed polyclonal hypergammaglobulinemia. LDH was 1,030.4 IU/L, total iron binding capacity 168 µg/dL, iron 13 µg/dL, and ferritin 2,050 ng/mL. A blood coagulation test showed increased prothrombin time, FDP, and D-dimer. The results of the serologic test were anti-HBs Ab (+), anti-HCV IgG Ab (+), EBV VCA-IgG (+), EBV VCA-IgM (-), EBNA (+), and EA (-). Both direct and indirect Coombs' tests were positive.
The peripheral blood smear showed normocytic and normochromic RBCs with poikilocytosis and anisochromia. Bone marrow aspirate showed hypercellular marrow with the infiltration of many small and medium sized T-lymphocytes without significant atypia, plasma cells, and many histiocytes. PCR analysis for the TCRγ gene rearrangement followed by SSCP showed a polyclonal pattern of the TCR gene.
A CT scan of the abdomen revealed a huge spleen of 17×14 cm in size and mild hepatomegaly. There were multiple subcapsular, low attenuating lesions. Multiple enlarged lymph nodes, measuring up to 3.7 cm in diameter, were found in the porta hepatis, portocaval, retropancreatic, and aortocaval regions.
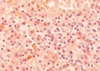
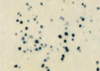
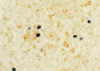
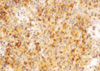
On the 6th day of admission, steroid was administered under the impression of autoimmune hemolytic anemia, but hematologic findings were not improved. Fever was persistent. On the 23rd hospital day, splenectomy with a paraaortic lymph node biopsy was done. The spleen was 18×16×7 cm and weighed 685 g. The cut surface showed multifocal infarcts involving about 30% of the parenchyma. Microscopically, red pulp was markedly expanded with atrophic white pulp. In the red pulp, there was diffuse infiltration of histiocytes showing erythrophagocytosis in the splenic cord and sinusoids. A few histiocytes phagocytosed platelets. Besides the histiocytes, many plasma cells had infiltrated the splenic cord. In some areas, extramedullary hematopoiesis and infarct of splenic tissue were observed. Paraaortic lymph node displayed loss of lymphoid follicles and paracortical expansion by diffuse infiltration of polyclonal plasma cells. The sinus contained a few erythrophagocytosing histiocytes. Immunostaining revealed many T-lymphocytic infiltrations with a predominance of CD4-positive cells in the red pulp as well as in the white pulp. Double procedure for CD3 immunohistochemistry and EBV in situ hybridization revealed numerous CD3-positive T lymphocytes harboring the EBV genome in their nuclei. CD56 was negative in those lymphocytes. A few B lymphocytes exhibited the EBV signal. The plasma cells in the bone marrow, spleen, and lymph nodes demonstrated a polyclonal light chain expression pattern.
After splenectomy, complete blood count was temporarily elevated, but fell again. One week after the surgery, the patient manifested body edema with ascites and petechiae of the skin. She also showed hyperbilirubinemia and elevated AST/ALT.
Under the diagnosis of chronic active EBV infection and DIC, acyclovir was administered to the patient and chemotherapy was planned. On the 34th hospital day, after the insertion of a Hickman catheter for chemotherapy, she complained of dyspnea. Chest radiography revealed bilateral pleural effusion and diffuse increase of hazziness of both lung fields. A bronchoscopic examination and chest radiographic findings indicated acute respiratory distress syndrome with diffuse alveolar hemorrhage. The cytomegalovirus was isolated in the culture of the bronchoalveolar larvage. The patient got worse rapidly. On the 65th hospital day, she was discharged with multiorgan failure by disseminated intravascular coagulation and septic shock (Figs. 1, 2, 3, 4, 5, 6).
DISCUSSION
EBV infection can cause a broad spectrum of diseases depending on the host's immune status. Primary infection in adolescent or early adult life can be manifested as a classic infectious mononucleosis characterized by fever, hepatosplenomegaly, lymphadenopathy, and an increase of activated CD8+ T lymphocytes in the peripheral blood. Fulminant EBV infection has been described under the name of fatal infectious mononucleosis and EBV-associated hemophagocytic syndrome in toddlers or young children. It is characterized by fever, hepatosplenomegaly, lymphadenopathy, hemophagocytosis in various tissues, and a tendency to pancytopenia, often leading to a fatal outcome (5, 6). Some cases of fatal infectious mononucleosis are associated with X-linked lymphoproliferative syndrome (7). Serologic profiles of those patients showed anti-VCA IgM, and IgG-positive, and anti-EBNA1-negative, which are diagnostic of a primary infection.
Some cases of fulminant EBV infection have been termed severe chronic active EBV infection (SCAEBV). The first patient described in 1978 was a 5-yr old girl with chronic infectious mononucleosis-like symptoms that had persisted for a long time. The patient also had extraordinarily elevated levels of IgG antibody titers against VCA and EA with EBNA-positive cells in circulation (8). Thereafter, some patients with a similar type of disease were reported, especially in the Asia. In Korea, only two cases have been described in the literature (9, 10). The clinical courses of these patients were fatal. The life-threatening complications included hemophagocytic syndrome, interstitial pneumonia, lymphoma, coronary artery aneurysm, or central nervous system involvement. In 1988, Straus et al. proposed the criteria of SCAEBV, which included severe illness lasting more than 6 months, histologic evidence of major organ involvement, abnormal EBV antibody titers, and increased quantities of EBV in affected tissues (11). Using the Straus et al. criteria as a guideline, Okano et al. reviewed the clinicopathologic and laboratory findings of 26 patients previously reported in the English literature, as well as their 10 cases, and emphasized that all of the patients had extremely high antibody titers against the EBV (12). However, the cases reported afterward did not always satisfy the proposed criteria by Straus et al. Some patients showed a duration of illness of less than 6 months, and other patients lacked abnormal patterns of EBV-related antibodies. In 1995, Ishihara et al. described the clinical and laboratory findings of 39 children with SCAEBV infection registered in Japan. In their series, past histories of hypersensitivity to mosquito bites were identified in 31.3% of patients, and high anti-EBV titers were noted in 28.2% of patients for anti-VCA IgG and in 57.1% of patients for anti-EA IgG (13). Recently, Kimura et al. analyzed 30 patients with SCAEBV and identified high EBV-related antibody titers in only one third of the patients. They also noted high viral loads detected by real-time quantitative PCR in the peripheral blood (more than 102.5 copies/µg DNA) in all of the patients (14). Based on these findings, Okano et al. suggested that virologic criteria for diagnosis may be altered to either or both extremely high antibody titers against EBV-replicative antigens and/or increased genome copies in tissues (15).
In the present patient, quantitative analysis of anti-EBV antibody titer and viral load quantitated by real-time PCR was not performed, but a pathologic examination identified many EBV-infected T-lymphocytes that had infiltrated in the spleen, lymph nodes, and bone marrow. Moreover, the patient showed fever, lymphadenopathy, and hepatosplenomegaly; and she pursued an aggressive fulminant clinical course with hemophagocytic syndrome, which is a typical clinical feature of severe chronic active EBV infection.
The short clinical course of 4 months from the onset of disease to death in the present patient does not satisfy the criteria of disease duration as given by previous authors, who emphasized an illness lasting more than 6 months' duration (12). However, a careful review of previous reports revealed that a few patients showing typical clinical and laboratory findings died of the disease at less than 2 months after the onset of the disease (16). Therefore, the present case suggests that the diagnosis of SCAEBV should be based on the constellation of clinical and laboratory findings and should not be limited by simple numerical criteria for disease duration or by laboratory parameters.
The majority of patients with SCAEBV at the onset of symptoms have been children and adolescents. Their average age was 8.3 yr. In the series by Okano et al. and Kimura et al., only 2 of 56 patients were over 30 yr-old at the onset of the disease, and there were no patients who were more than 60 yr-old (12, 14). In consideration of serologic profiles showing detectable IgG antibody titers to EBNA along with high titers of IgG anti-VCA and anti-EA antibodies, most young patients with CAEBV appeared to have had active EBV infections and had already suffered from primary EBV infection at the onset of CAEBV. Many cases of CAEBV in older patients may occur with the reactivation or reinfection of the EBV.
In the present case, a double procedure for immunohistochemistry and EBER in situ hybridization revealed that numerous CD3-positive T lymphocytes had harbored the EBV genome in their nuclei. Unlike classical infectious mononucleosis in which EBV infects B cells via the EBV receptor on the cell surface, which are eliminated by EBV-antigen directed cytotoxic T cells, SCAEBV is characterized by persistent EBV infection into T or NK cells and the proliferation of these EBV-positive populations for long periods (17, 18). A recent study by Kimura et al. demonstrated the clinical and virologic differences between the two subgroups of SCAEBV infecting the T or NK cell. The T cell type CAEBV tended to show high titers of anti-EBV-related antibodies and a tendency of early mortality compared to the NK cell type CAEBV (14). Infection of the T-cell, instead the B-cell, by EBV is also observed in EBV-associated hemophagocytic syndrome (EBV-HPS), which can be associated with acute EBV infection. In a study by Kasahara et al., 4 patients with acute EBV-HPS showed predominant CD8+ EBV infected T cells; whereas dominant EBV-infected cells in patients with CAEBV were CD8+ T cells or CD4+ T cells or NK. The difference of predominantly infected cells between acute EBV-HPS and CAEBV might be ascribed to the differences in the time of analysis after the onset of disease; both the CD4+ and the CD8+ T cells became infected with the virus as the disease progressed (4).
Infectious mononucleosis is common in western countries, but the majority of the fulminant EBV infections including EBV-HPS and SCAEBV infection has been reported in Asia, including Japan and Taiwan. This suggests a certain racial difference in the immunological function which controls EBV infection. Although the patients reported hitherto had no proven immune defects, it has been suggested that a defect in the gene essential for regulating lymphocyte activation and proliferation may exist. Another suggestion is that the virus may escape from the immune surveillance by the host because of the mutation of the viral genes. In the present patient, chronic hepatitis C virus infection and INF-α treatment might have caused the immune dysfunction; however, the clinical course does not support a causal relationship between HCV infection and SCAEBV infection.
In summary, SCAEBV infection is a fulminant disease caused by EBV. Although high antibody titers for EBV antigens and duration of illness are included in the diagnostic criteria, flexible application of those criteria is necessary in cases showing typical clinical and pathologic findings.

 XML Download
XML Download