

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Adult onset Still's disease (AOSD) is a chronic systemic inflammatory disorder of unknown etiology, and its major clinical manifestations include high spiking fever, polyarthralgia, salmon colored evanescent rash, and neutrophilic leukocytosis. Life threatening conditions such as hepatic involvement, cardiac tamponade, disseminated intravascular coagulation (DIC), respiratory distress syndrome or pancytopenia were occasionally developed in the course of AOSD (1), and in some cases were often associated with hemophagocytic syndrome (HS) (2-4). Most patients with HS have rapid and fatal outcomes unless the diagnosis is made early and followed by prompt therapeutic intervention. There have been some fatalities due to a delayed recognition in reported AOSD patients with HS, because many of the symptoms of HS are overlapped with those of AOSD and mimic sepsis. We report a successfully treated case of a patient with AOSD in which DIC and multiple organ dysfunctions were presumed.
CASE REPORT
A 49-yr-old woman with a 7-yr history of AOSD was admitted to emergency room due to deterioration of consciousness with a few hours of duration. Four years before the admission, she had been admitted for high spiking fever, evanescent morbilliform rash, polyarthritis, neutrophilic leukocytosis and hyperferritinemia (21,239 ng/mL). At that time she had been diagnosed as a flare-up of AOSD by Yamaguchi's criteria (5), and the course of the disease had been improved with moderate dose of prednisolone (PSL).
Since the first admission, she had had recurrent episodes of fever, rash and polyarthritis mimicked rheumatoid arthritis. Her symptoms were dependent on PSL, and the course of the disease did not change in spite of concurrent treatment with sulfasalazine, methotrexate, bucillamine, azathioprine, cyclosporine and cyclophosphamide. One month prior to the admission, she had been treated with famciclovir (750 mg/day) for 1 week due to acute herpes zoster rash on left forearm. Until 4 days before the admission, she had received PSL (5 mg/day), hydroxychloroquine (400 mg/day) and sulindac (200 mg/day) for a month, as well as elemental iron (80 mg/day) for 8 months because of iron deficiency anemia.
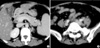
On admission (4th day after the onset), she appeared acutely ill with a confused mental status. Her vital signs were the blood pressure of 90/60 mmHg, the heart rate of 100/min, the temperature of 39.0℃, and the respiratory rate of 30/min. Physical examination revealed facial rash without coalescence, icteric sclera, dehydrated tongue, equivocal neck stiffness, splenomegaly, purpuras over the limbs, scabs of zoster on left forearm and severe tenderness in the right upper and lower quadrant of the abdomen with positive Murphy's sign. There was no definite abdominal rigidity or palpable lymphadenopathy. Initial laboratory results were as follows: WBC 7,100/µL (neutrophil 62%, bands 10%, lymphocyte 15%, monocyte 12%), hemoglobin 9.5g/dL, mean corpuscular volume 74.2 fL (normal 79-95), platelet 17,000/µL, reticulocytes 0.2%, iron 164 µg/dL (normal 50-150), TIBC 218 µg/dL (normal 250-400), ferritin>1,831 ng/mL (normal 10-291), ESR 10 mm/hr, C-reactive protein 16.5 mg/dL (normal 0.1-0.8), total bilirubin 3.7 mg/dL, direct bilirubin 2.8 mg/dL, AST 453 U/L, ALT 154 U/L, ALP 356 U/L, LDH 2,350 U/L, CK 1,547 U/L, BUN 41 mg/dL, creatinine 2.3 mg/dL, total cholesterol 79 mg/dL (normal 130-250), triglyceride 335 mg/dL (normal 50-150), HDL-cholesterol 13 mg/dL (normal 30-70), C3 88.8 mg/dL (normal 79-152) and C4 13.8 mg/dL (normal 16-38). Coagulation tests revealed the following results: PT 18 s (control 12), aPTT 98 s (normal 23-39), fibrinogen 52 mg/dL (normal 190-430), FDP 40 µg/mL (normal<10), antithrombin III 33.9 % (normal 80-120) and D-dimer 4 mg/L (normal<0.3). Direct and indirect Coomb's tests were negative. Blood smear revealed polychromasia, combined with normocytic and microcytic anemia and severe thrombocytopenia. Antibodies to nuclear antigens, dsDNA, cardiolipin, VDRL and rheumatoid factor were negative. Urine examination revealed protein 1.41 g/day with granular casts, fractional excretion of sodium measuring 1% and urine sodium concentration 21 mmol/L. Antistreptolysin O and Widal test were normal. Hepatitis B surface antigen, hepatitis C virus, and HIV antibodies were negative. Serological tests for varicella-zoster virus (VZV), Epstein-Barr virus, cytomegalovirus and herpes simplex viruses revealed no signs of recent infections. Chest radiograph and electrocardiogram appeared normal. Abdominal computed tomography (CT) images showed acute acalculous cholecystitis, ileocolitis (Fig. 1), hepatosplenomegaly, a small amount of right pleural effusion and ascites. The patient had no history of recurrent episode of abdominal pain or diarrhea. Bone marrow (BM) biopsy and cerebrospinal fluid examination were not performed due to hemorrhagic diathesis.
The combination of anemia, thrombocytopenia, coagulopathy, DIC, hepatic and renal dysfunctions, neurological symptoms, and capillary leakage signs was compatible with severe systemic inflammatory response syndrome. A differential diagnosis of hemophagocytic syndrome and septic DIC was considered. Fluid and electrolytes replacement, fresh frozen plasma and platelet concentrates transfusions were immediately initiated. Empirical antibiotics (ceftriaxone, 3 g/day) and intravenous immunoglobulin (IVIG, 400 mg/kg/day) therapy were also initiated. On the next day after the admission, confused mentality diminished, and serum creatinine level was normalized. Negative results for blood, throat, urine and stool cultures were noted. On the basis of patient's clinical presentations, marked elevated ferritin, hypertriglyceridemia, and no evidence of infectious etiology, a presumptive diagnosis of reactive hemophagocytic syndrome (RHS) associated with AOSD was made. Intravenous pulsed methylprednisolone (Methyl-PSL, 1 g/day) and oral cyclosporine A (CsA, 2 mg/kg/day) were added. Two day after the admission, fever was disappeared rapidly, and abdominal tenderness was also decreased. On fourth day of the admission, high fever and neurological dysfunctions such as confusion, irritability, disorientation abruptly recurred, and abdominal tenderness re-aggravated despite treatment with IVIG (for 3 days), pulsed methyl-PSL (for 3 days) followed by methyl-PSL 62.5 mg/day, continuous oral CsA and blood component replacements. Although levels of transaminase and LDH were gradually decreased, pancytopenia and DIC progressed. On the sixth day of the admission, we increased the dosage of oral CsA from 2 mg/kg/day to 3 mg/kg/day. Fever and confusion were disappeared dramatically within a day. She no longer needed any transfusion, with pancytopenia and DIC rapidly improving. Subsequently, abdominal tenderness also disappeared within 3 days. On day 42 of the hospitalization, all laboratory abnormalities except lipid profile returned to normal (Fig. 2). Previous radiological abnormalities were also normalized on follow-up study. CsA administration was stopped, and PSL was tapered. She has remained clinically well with low-dose PSL alone for over one year.
DISCUSSION
HS is an uncommon disorder characterized by inappropriate systemic proliferation of benign histiocytes throughout the reticuloendothelial system and hemophagocytosis. It has been suggested to be caused by extensively activated T cells (Th1 cells) and macrophages, and subsequent overproduction of cytokines such as IL-1, IL-6 and IFN-γ (6). It can occur as a primary hemophagocytic lymphohistiocytosis, but more commonly is secondary (also called RHS) to a variety of infections, neoplasms, drugs, autoimmune diseases or various immunodeficiencies. Among the rheumatic disease, systemic onset juvenile rheumatoid arthritis (SOJRA) is most often associated with HS, and it has been named macrophage activation syndrome (MAS) (7). As AOSD is similar to Still's disease, it seems to be prone to developing reactive HS in adults (8).
Although we did not identify tissue demonstration of hemophagocytosis in our case, typical clinical and laboratory findings of defining HS, such as the sudden onset of non-remitting high fever, change in mental state, hepatosplenomegaly, serositis, cytopenia, elevated serum transaminase, hyperbilirubinemia, high concentrations of LDH and triglyceride, coagulopathy with prolonged PT and aPTT, hypofibrinogenemia, the presence of fibrin degradation products, hyperferritinemia, complete resolutions of all symptoms and abnormal findings by immunosuppressive therapy, can reasonably establish the diagnosis of HS (6, 8-10). In fact, the absence of histological confirmation of hemophagocytsis must not be allowed to delay treatment, which is urgently required. Our chronic AOSD patient with HS became acutely ill, and the clinical picture mimicked septic DIC or a flare-up of AOSD with complicating DIC. However, patterns of non-remitting fever and purpuritic or petechial rash were different from the remitting high-spiking fever and evanescent maculopapular rash of AOSD. The most dramatic clue was the fall in the ESR inspite of both the face of a worsening clinical situation and the very high levels of ferritin. Low ESR is a strikingly unusual feature in active phase of inflammatory disease, and thought be related to hypofibrinogenemia. Hyperferritinemia is an important laboratory hallmark for diagnosis of HS and disease activity of AOSD. Patients with HS showed markedly elevated serum ferritin levels ranging from 1,000 to 250,000 ng/mL (2, 3). In the appropriate clinical setting, a serum ferritin level exceeding 1,000 ng/mL may assist in establishing the diagnosis of HS (6).
The pathognomic feature of the HS is seen on BM examination, which reveals numerous well-differentiated macrphages actively phagocytosing hematopoietic cells. Association between massive hyperferritinemia and hemophagocytosis is strong, but BM specimen may be inconclusive in case of early sampling time or hemophagocytosis sparing bone marrow (8, 11, 12). However, BM analysis is recommended in all patients with hyperferritinemias, not only for diagnostic accuracy but also given the high percentage of underlying malignancies such as malignant histiocytosis or lymphoma (13, 14). In our patient, the dramatic restoration of general condition by immunosuppressive treatment without a specific chemotherapy would support the unlikeness of malignancy associated HS inspite of omitting BM examination.
HS commonly occurs in patients with pre-existing immunological abnormalities due to various infections (15). In this case, herpes zoster preceded HS a month before the admission. Presumed acute acalculous cholecystitis and ileocolitis were found in CT images on admission. Therefore, we suspected infection associated hemophagocytic syndrome at first. Viral infection may be a delayed trigger of HS whether antiviral agent was used or not (10). In fact, viral-associated HS frequently resulted in severe pancytopenia, DIC and fulminant clinical course due to multiple organ failure. Enterocolitis and acute non-calculous cholecystitis can be concurrently caused by same pathogen (16). However, no active infection of viruses including VZV, bacteria, fungi, and parasites were detected in our case. Actually, it is difficult to confirm the absence of any infectious agents thoroughly. Although we treated this patient with a moderate dose of single broad-spectrum antibiotic as well as immunosuppressive agents, the lack of aggravation or relapse of infection under aggressive immunosuppression would also favor the unlikeness of underlying non-viral infection.
On the one hand, diffuse wall thickenings in colon, cecum and terminal ileum observed on CT suggests that inflammatory bowel disease such as Crohn's disease cannot be ruled out. HS associated with inflammatory bowel disease (IBD) is very rare and we are aware of only one previous case report of fulminant ulcerative colitis associated HS (17). However, our patient had no previous history of recurrent episodes of abdominal pain or diarrhea at presentation of HS. After recovery, the patient has not shown a sign or symptom of IBD yet. Therefore, it is possible to consider the bowel lesion as a result of inflammatory response by hypercytokinemia or tissue infiltrations by histiocytes associated with HS rather than a feature of IBD itself. The classical feature of acute acalculous cholecytitis revealed in our case is atypical, but patients with severe AOSD or HS often present with an acute abdomen. Abdominal pain is supposed to be due to peritoneal inflammation, acute enlargement of mesenteric lymph node, or functional obstruction of the intestine (1). In our case, there is a possibility that acute serositis mimics acute cholecystitis. We think that it is important to recognize this as a clinical feature of RHS with AOSD in order to avoid unnecessary surgical exploration. A number of triggers for MAS in SOJRA have been proposed, including aspirin or other non-steroidal anti-inflammatory drug toxicity, a second injection of gold salts, sulfasalazine and methotrexate therapy, as well as a viral infection (7). In our case, the patient had received treatment with sulindac, hydroxychloroquine and oral ferrous sulfate at the onset of RHS, but there have been no reports of RHS related to these drugs. However, a case of acute hepatitis in AOSD apparently resulting from oral iron substitution was recently reported, and there was a suggestion that the iron exacerbated the macrophage hyperactivity in active phase of AOSD (18). In our case, RHS did not develop during the long-term iron supplement period for more than 8 months despite several flare-ups of AOSD. Therefore, RHS triggered by iron would be less likely.
Because RHS may have a fatal outcome, prompt recognition and treatment are of the uppermost importance. The treatment strategy for RHS is usually based on the parenteral administration of high doses of corticosteroids. However, corticosteroid is effective in most but not all cases, and the use of high-dose IVIG, cyclophosphamide, plasma exchange, and etoposide has provided conflicting results. TNF-α blocker may also be an effective therapeutic agent in conjunction with corticosteroid, and a steroid-sparing agent (19). CsA proved effective in treating severe or corticosteroid-resistant HS (20, 21). In some patients with MAS, the introduction of this drug leads a dramatic effect on the process of the disease, leading to resolution of the fever and improvement of the laboratory abnormalities within 12 to 24 hr (20). Although the exact mechanism by which CsA achieves immunosuppression is unknown, it is believed to exert its major effects by the suppression of the early steps in T-cell activation, leading to failure to activate the transcription of early genes such as those encoding for cytokines (22). CsA has also been shown to affect macrophage production of IL-6, IL-1 and TNF-α and to inhibit the expression of inducible nitric oxide synthetase and cyclooxygenase-2 in macrophages (23-25). CsA also inhibits the expression of key cell surface co-stimulatory molecules, thus altering the antigen-presenting function of dendritic cells for T cell activation (26).
To our knowledge, this is the first case of AOSD associated HS masquerading as acute acalculous cholecystitis and ileocolitis. We suggest that CsA can be considered as first-line treatment with steroids in life threatening HS such as DIC and multiple organ dysfunctions.

 XML Download
XML Download