

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
X-linked severe combined immunodeficiency (X-SCID) is a rare, life-threatening disease that is characterized by marked impairment of both cellular and humoral immunity (1). Human X-SCID occurs in as many as 50% of patients with primary SCID; patients have complete or marked deficiency of T cells but carry a normal or slightly increased number of B cells, despite a deficiency in specific antibody responses (2). Infants with SCID have no tonsils, rarely have detectable peripheral lymph nodes, and have a small, poorly differentiated thymus gland. Without bone marrow transplantation (BMT), affected patients suffer severe and persistent infections, often with opportunistic pathogens, and generally die in infancy.
The abnormal gene was mapped to the Xq13 region, and later identified as the gene encoding the common gamma (γc) chain (3, 4) that is common to the cell-surface receptors for six interleukin (IL) molecules (IL-2, 4, 7, 9, 15, and 21) (2-5). Based on the amino acid sequence, the complete cDNA clone encoding the 64-kDa molecule was isolated and demonstrated to be the cognate IL-2Rγ chain (6, 7). The intracellular portion of the γc chain is known to interact with Janus kinase 3 (Jak3), a signaling kinase that cooperates with other Jak and STAT proteins in a complex signal transduction array (8, 9).
Various mutations in the γc chain gene have been reported in patients with X-SCID (10-15). However, little is known about the clinical and immunological characteristics, or the genetic polymorphism in Korean patients with X-SCID. Here, we report the mutation identified in one Korean family with X-SCID, along with prenatal diagnosis of the disease by RFLP analysis. To our knowledge, this is the first report of the molecular diagnosis of X-SCID in a Korean patient.
CASE REPORT
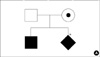
A 13-month-old boy was transferred to the Department of Pediatrics, Chonnam National University Hospital, because of refractory, progressive pneumonia, and nodular skin lesions. From the age of 7 months, he frequently visited his primary physician because of recurrent upper respiratory infections. The birth history was not contributory, but there was an infant death history of unknown etiology for his uncle on the mother's side. The pedigree of the family is shown in Fig. 1A. He received routine immunizations until 6 months of age, including BCG.
On admission, he was immediately intubated and maintained with an artificial ventilator because of severe, bilateral pneumonia causing respiratory failure. The blood counts on admission were: white blood cell, 45,100/µL (neutrophil 75.5%, lymphocyte 15.1%); hemoglobin, 14.4 g/dL; platelets 209,000/µL. His serum immunoglobulin profile was: IgG, 27.0 mg/dL (normal range, 345-1,236 mg/dL); IgM, 50.4 mg/dL (41-173 mg/dL); IgA, 22.9 mg/dL (14-159 mg/dL). Flow cytometric analysis of lymphocyte immunophenotype was: CD3 T cells, 2.5% (normal range, 60-85%); CD19 B cells, 95.4% (8-20%); CD16/CD56 natural killer cells, 0.5%. He was thus shown to have T-B+NK- SCID.
On physical examination, he had no BCG scar, and biopsy on the nodular skin lesion was later found to be a tuberculous granuloma. He was treated with broad-spectrum antibiotics, anti-tuberculosis agents, prophylactic antifungal agents, and intravenous globulins. He gradually recovered and was weaned off the ventilator on the 63rd hospital day. The recent blood counts without infection were: 5,300/µL (neutrophils, 58.5%; lymphocytes, 27.4%); hemoglobin, 12.5 g/dL; platelets 265,000/µL.
Blood samples were obtained from the patient and various family members with written informed consent. Heparinized venous blood samples from the patient and his family members were fractionated on a Ficoll-Hypaque gradient to isolate peripheral blood mononuclear cells (PBMCs). PBMCs were washed twice in PBS and 0.02% NaN3, and analyzed by flow cytometry for γc chain expression using the PE-conjugated rat anti-human γc chain monoclonal antibody TUGh4 (Beckton-Dickinson, Mountain View, CA, U.S.A.). Incubation with unlabeled TUGh4 before the addition of labeled TUGh4 provided the background control staining. The method for immunofluorescence staining was the same as that described previously (16). As shown in Fig. 1B, PBMCs from the mother showed γc chain expression on both CD3- and CD19-positive populations. In contrast, cells from the patient showed a very depressed γc chain expression on CD3- and CD19-positive cells, suggesting both T and B cells from the patient did not express γc chain on the cell surface (Fig. 1B).
The eight exons of the γc chain and surrounding genomic sequences were amplified from genomic DNA as described previously (12). As shown in Fig. 2A, we found a single base substitution (C690T) at exon 5, resulting in an amino acid change at codon 226 (R226C) in the patient. His mother was found to have one mutant and one normal allele, indicating that she was a heterozygous carrier. As the C-to-T substitution predicted the creation of a new AluI restriction endonuclease site, the sequencing results were further confirmed by restriction fragment length polymorphism (RFLP) analysis (Fig. 2B). The patient showed DNA cleavage with AluI (bands of 179 and 133 bp), while the normal control was resistant to AluI. The mother possessed an uncleaved (312 bp) and two smaller bands identical to those in the patient, implying heterozygosity.
By RFLP analysis, we were able to perform a prenatal diagnosis on a male fetus. At the 24th week of gestation, 3 mL of umbilical cord blood was sampled percutaneously, and analyzed for lymphocyte phenotyping. The fetus had the same T-B+NK- phenotype (CD3 T cells, 1.2%; CD19 B cells, 78.6%; CD16/CD56 natural killer cells, 1.2%). Fetal genomic DNA isolated from amniotic cells was analyzed by RFLP for exon 5. As shown in Fig. 2C, the fetal DNA showed the same cleaved DNA pattern as the patient, while the healthy control DNA showed the normal pattern. RFLP analysis demonstrated that the fetus had the same mutation as his brother, and the pregnancy was terminated after lengthy counseling. The patient is now being cared for at our outpatient clinic, awaiting a stem cell transplant from an alternative donor, because he does not have an HLA-matched sibling.
DISCUSSION
Here, we report the first mutation analysis of the γc chain gene in a presumed X-SCID patient from a Korean family. The patient had a single point mutation (C690T) at exon 5, resulting in an amino acid change at codon 226 (R226C). The National Institutes of Health database of γc chain mutations lists 264 mutation entries, comprising 169 unique molecular events (http://www.nhgri.nih.gov/DIR/GMBB/SCID/IL2RGbase.html). Mutations have been identified throughout all eight exons of the γc chain, the majority of which are missense mutations, nonsense mutations, or splicing defects that give rise to aberrant transcripts. The missense mutation found in this study is not novel. The available data suggest that the codon 690 may be a hot spot for γc chain gene mutation, as reported in several studies (10, 12-15). We did not examine the stability of mRNA or protein expression of the γc chain in this study, but our previous study demonstrated that the γc chain containing the point mutation was not expressed on the cell surface because it was trapped in the Golgi apparatus and failed to be transported to the cell surface (12). Further identification of the locations and types of mutations in the γc chain gene may lead to a better understanding of the functional domains of the protein.
In addition, the patient showed a very little expression of γc chain on both B and T cells. In a previous study, cell-surface staining of patient-derived B cell lines with an anti-γc chain antibody showed no γc chain staining in 47% and trace amounts in 32%, but normal intensity of staining in 21% of patients (11). In another study, rapid protein analysis with flow cytometry and immunoblotting were shown to be useful for diagnosis of X-SCID and JAK3-deficient SCID. Although the patients had mutations in the γc chain gene, some had trace or normal levels of γc chain expression. Thus, these proteinbased assays have led to rapid molecular diagnoses of X-SCID that have subsequently been confirmed by genetic analysis.
The male X-SCID patient in the present study had a classical, severe phenotype, and laboratory data showed low numbers of T cells, relatively well-preserved B cells, and reduced NK cell numbers. X-SCID is a disease that is characterized by severe lymphopenia and recurring persistent infections in the first months of life (17). A human Jak3-deficiency disease has similar clinical features to X-SCID (18), and the γc chain and Jak3 signaling pathway have been suggested to be crucial for T cell development, and to contribute to the X-SCID phenotype (18). In addition, gene disruption of either IL-7 (19) or the IL-7R subunit (20) has been shown to lead to severe developmental perturbations. Thus, signaling defects in the IL-7/IL-7R system resulting from changes in the γc chain may account for the developmental anomalies that lead to X-SCID.
We also performed a prenatal diagnosis for this family, based on our genetic findings. Most of the primary immunodeficiency diseases can be diagnosed by means of screening for lymphopenia or for T cell deficiency in cord blood at birth. In addition, fully defining the molecular defects of patients is essential for genetic counseling of family members and prenatal diagnosis.
In conclusion, we described here the molecular and cellular identification of an X-SCID patient in a Korean family. Our data emphasize that flow cytometric analysis is an important and powerful assay that can contribute to diagnosis of X-SCID. Further studies of X-SCID mutations should be compiled and analyzed to provide data to clarify the correlation between the severity of the disease and the types and locations of the mutations in X-SCID patients.

 XML Download
XML Download