

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Kinesin proteins make up a large superfamily of molecular motors, known as kinesin superfamily proteins (KIFs). The first kinesin was characterized by Vale et al. in 1985 as a cytosolic, microtubule-stimulated ATPase responsible for the directed, ATP-dependent-vesicle movement within squid axons (1). At the time, two motors, kinesin and dynein, seemed almost sufficient to explain directional motility events on polar microtubule tracks in the cell. Nonetheless, shortly after, the tip of the iceberg began to emerge with the identification of proteins containing a common domain. This domain, called the "motor domain", confers on these proteins the essential property of moving on microtubules, using the energy derived from ATP hydrolysis. Since then, the identification of new proteins belonging to the kinesin superfamily of microtubule-dependent motors has gone at such a pace that nowadays more than 200 entries with motor domain sequences are deposited in the database.
Until now, more than 45 KIFs have been reported in mouse and human genomes, which have two major functions (2). KIFs participate in the movement of cargoes to specific destinations along cytoskeletal tracks of microtubules (3). Additionally, KIFs are also involved in the organization of chromosomal and mitotic spindle movements (4, 5). Structurally, KIFs consist of two functional domains: a motor domain that reversibly binds to the cytoskeleton; and the rest of the molecule, often referred to as the tail that interacts with cargo directly (2, 3). We still do not understand how a specific kinesin is linked to its cargo to ensure its delivery to the appropriate location. Recently, several kinesin binding proteins have been identified by yeast two-hybrid, genetic studies, and biochemical analyses, and their roles in the cargo transport are now being elucidated. Many cell biological approaches have demonstrated that binding proteins and the motor function impact all dynamic aspects of the generation and maintenance of cell polarity and cell motility. Here we discuss the functions of KIFs, cargo binding proteins and kinesin motor-based human diseases.
KINESIN STRUCTURE AND CLASSIFICATION
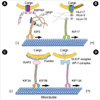
The original 'conventional' kinesin (alias KIF5/kinesin-I) was shown to be a tetrameric protein composed of two heavy chains (110-120 kDa) and two light chains (60-70 kDa). Electron microscopy, protease sensitivity and primary sequence analysis showed that the kinesin heavy chain (KHC) is composed of three domains: N-terminal head domain, α-helical stalk domain and C-terminal tail domain (6, 7). The globular N-terminal head domain (residues 1-325) contains the adenosine triphosphate (ATP)-binding motif and a microtubule-binding domain. It is attached via 50 amino acid neck regions to an extended α-helical stalk (residues 375-800), which forms a coiled-coil upon dimerization with a second heavy chain. The C-terminal tail domain (residues 800-963) is globular and interacts with the kinesin light chains (KLC) (4). Outside the motor domain, KIFs show few similarities to each other. Recently, it has been clearly shown that several KIFs attach to specific cargoes through interactions with adaptor proteins in the C-terminal tail region. Since the characterization of the first kinesin, many related proteins have been discovered, including conventional kinesins as well as proteins belonging to the growing kinesin superfamily proteins. A database search of the total human genome indicated that there are a total of 45 KIFs (2).
Three major types of KIFs have been identified according to the position of the motor domain: NH2-terminal motor domain type (N-type), middle motor domain type (M-type), and COOH-terminal motor domain type (C-type) (2, 3). N-type kinesins (KIF1, KIF3, KIF4, KIF5, KIF10, KIF12, KIF 13, KIF14, KIF15, KIF16, KIF17, KIF18, KIF20, KIF23, KIF26) and M-type kinesin (KIF2) move exclusively towards the plus-ends of microtubules like the cell periphery or synapse terminal, whereas C-type kinesins (KIFC1, KIFC2, KIFC3) move towards the minus-end (retrograde, or toward the nucleus, to a microtubule-organizing center, MTOC). Rotary shadowing and biochemical analysis shows that there are five types of conformation for kinesin superfamily protein: homo-, and heterodimers, heterotrimers (KIF3 containing two distinct heavy chains, KIF3A and KIF3B, and one light chain, KAP3), heterotetrameric (KIF5) and monomers (KIF1) (3) (Fig. 1, 1).
CYTOPLASMIC KINESIN PROTEIN AND ITS INTERACTION
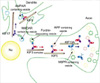
Although the interaction of motors and cargoes are only poorly understood, this is an area of rapid progress (8). Schliwa's group searched for functional KHC domains using KHC deletion mutant cDNAs expressed as transgenes and evaluated their ability to rescue the deduced growth rate of a KHC-deficient strain of Neurospora. In Neurospora, the tail coiled-coil domain (820-918 amino acids) of KHC is essential for targeting and secretion to small vesicles at the hyphal tip (9). Hirokawa's group showed that an AMPA (α-amino-3-hydroxy-5-methylisoxazole-4-propionate) receptor subunit-glutamate-receptor-interacting protein (GRIP1), one of the multi-PDZ-domain proteins at the synapse junction (10, 11), can directly interact with this tail coiled-coil domain of KHC and steer to dendrites as a motor for AMPA receptor (12) (Fig. 2A). The region between the sixth and seventh PDZ domain of GRIP1 binds to a region of KHC that overlaps the tail coiledcoil domain defined in Neurospora. This interaction was further confirmed by the observation that KHC and GRIP1 are present on the same vesicles within the dendrites of cultured hippocampus neurons and co-immunoprecitate in the vesicle fractions. In addition, both gene targeting and dominant negative experiments of KHC resulted in abnormal localization of GRIP1 and GluR2. In GRIP1 expressing neuronal cells, KIF5 carries GRIP1 associated vesicles towards dendrites; on the other hand, in cells expressing JSAP1/JIP3, KIF5 transports JSAP1 associated vesicles towards axons (12, 13). A simple explanation of this phenomenon is that binding proteins determine the general direction of the traffic and also that KIF family members can recognize multiple binding proteins or cargoes.
A newly identified kinesin-like protein, KIF17, interacts with the PDZ domain of sorting protein mLin-10 (Mint1/X11), which is a component of a large complex that includes mLin-2 (CASK), mLin-7 (Velis) and the transmembrane protein NR2B, a subunit of the N-methyl-D-aspartate (NMDA) receptor (14). In the experiments of Hirokawa's group, immunoprecipitation of KIF17 brought down the LIN complex as well as the NMDA-receptor. In vitro vesicle motility experiments and dominant inhibitory KIF17 constructs inhibit nucleotide-dependent microtubule binding of NR2B-containing vesicles isolated from the brain and KIF17 promotes the motility of isolated NR2B-containing vesicles along microtubules (Fig. 2B). Also, cellular knockdown or functional blockage of KIF17 expression using antisense oligonucleotides decrease NR2B and mLin10 expression. Moreover, increase in the expression level of NR2B by exposure to the NMDAR antagonist increases the expression level of KIF17 (15).
Another KIF family member, KIF3 is one of the most abundantly and ubiquitously expressed KIFs and predominantly found in the nervous system (16). It is composed of a heterotrimeric complex formed with KIF3A, either KIF3B or KIF 3C, and an associated protein, KAP3 (17). In a yeast two-hybrid binding assay, it was revealed that the KIF3 motor and non-erythroid spectrin, termed fodrin, directly interacts through the non-motor subunit of KIF3 and KAP3 (Fig. 2C). Immunoprecipitation and immunoelectron microscopy further confirmed the co-localization of fodrin and KIF3 on the same vesicles, reinforcing the evidence that the cargo of the KIF3 motor consists of fodrin-associating vesicles. In addition, a pulse-labeling study has implied partial co-migration of both molecules as fast flow components (18).
Using affinity purification, velocity gradient, and direct in vitro binding assays, another novel kinesin-like protein called KIF13A was found to interact with the AP-1 clathrin-associated adaptor complex, which helps mediate the transport of coated vesicles from the trans-Golgi network (TGN) to the plasma membrane (19). Also, using yeast two-hybrid, it was verified that the C-terminal tail domain of KIF13A binds to the β1-adaptin subunit of the AP-1 complex (20) (Fig. 2D). Hirokawa's group also showed that KIF13A, AP-1 and the mannose-6-phosphate receptor (M6PR) are co-immunoprecipitated from cell lysates, and overexpression of the KIF13A tail domain decreases the amount of M6PR at the plasma membrane. These data demonstrate a link between the AP-1 complex and the cytoskeleton that allows KIF13A targeting of vesicles containing M6PR from the TGN to the plasma membrane (Fig. 3).
As mentioned above, there are a total of 45 KIFs in the human (2), which are responsible for many of the major microtubule-dependent transport pathways in neuronal and non-neuronal cells (21). At least several hundred different transport complexes and membrane-bound organelles can travel along microtubules. The diversity of these players, such as the motor protein, a cargo-bound receptor, and accessory components may explain how a limited number of kinesins do their work for the wide variety of cellular transport tasks. Despite the current knowledge of kinesin binding proteins detailed above, there are no known receptors (adaptor proteins) for cargoes such as synaptic vesicles, mitochondria, lysosomes and mRNA complexes, which are also dependent on kinesin transport.
HOW COULD MOTORS BE INVOLVED IN HUMAN DISEASES?
CMT and neurodegenerative diseases
Through the study of molecular motors, axonal transport has been proven to be involved in Charcot-Marie-Tooth disease (CMT) 2A pathogenesis (22). CMT is classified into type I and type II by using motor nerve conduction velocity as a marker of myelin degeneration (23, 24). KIF1Bβ is an isoform of the conventional mitochondrial motor KIF1B. It is widely distributed both in the cell bodies and axons of neurons, and its tail domain has a 61% amino acid homology to that of KIF1A (25). CMT2A, an autosomal dominant subtype of type II CMT, was mapped to human chromosome 1p35-36. The human KIF1B locus is closely linked to the CMT2A locus (22). Hirokawa's group analyzed the KIF1B locus in CMT2A patients and discovered a Q98L missense mutation in the ATP binding consensus sequence of the motor domain. KIF1B knockout mice die at birth from apnea due to nervous system defects. KIF1B heterozygotes have a defect in synaptic vesicle precursor transport and suffer from progressive muscle weakness with a motor nerve conduction velocity within the normal range, resembling the symptoms of CMT2. A cell biological approach demonstrated that KIF1Bβ co-fractionates with membranous organelles containing synaptic vesicle protein and is co-localized with synaptic vesicle protein on vesicle membranes. GST-pull down and immunoprecipitation assays with a membrane vesicle fraction of mouse brain also showed that KIF1Bβ is associated with vesicles containing synaptic vesicle proteins such as synaptotagmins, synaptophysin and SV2. Accordingly, in CMT2A patients and kif1B+/- mice, haploinsufficiency of the KIF1Bβ motor results in a deficiency of the cargo proteins being transported by this motor, including synaptic vesicle proteins in nerve axons and endings, brings about progressive dysfunction of peripheral neurons (22).
KIF1A, a murine homologue of Unc-104 in Caenorhabditis elegans, is a unique monomeric neuron-specific microtubule plus end-directed motor (26). KIF1A mediates the transport of a synaptic vesicle precursor and is essential for the viability, maintenance and function of neurons, particularly of mature neurons (27). In some neurodegenerative diseases, such as senile dementia, neuronal cell death caused by defects in the transport of synaptic vesicle precursors by KIF1A may be involved.
Polycystic kidney disease and Kartagener's syndrome
Polycystic kidney disease is a common genetic disorder that is characterized by the accumulation of fluid-filled cysts in the kidney, liver and other organs (28, 29). Several proteins that are encoded by genes associated with polycystic kidney disease have been identified in primary cilia. Also, recent observations suggest that abnormalities of primary cilia play a role in the cyst formation. Primary cilia are hair-like organelles that consist of an axoneme containing nine peripheral pairs of microtubules surrounded by a ciliary membrane (30). The synthesis of primary cilia involves a process known as intraflagellar transport, in which large particles containing protein cargo are transported along the ciliary axoneme (31). The anterograde movement of proteins along the axoneme is mediated by KIF3. In experiments by Hirokawa's and Goldstein's groups, mutations in the gene encoding the KIF3A or KIF3B subunits in mice cause an embryonic lethal phenotype (32-34). The nodal cilia, in which the heterotrimeric motor KIF3 is localized, rotate to generate a unidirectional flow of extra-embryonic fluid (nodal flow), which could fundamentally control left-right determination (32). In the null mutants of KIF3, there are no nodal flows. Thus, KIF3 is essential for development of the left-right axis determination due to its involvement in intraciliary transport of materials for ciliogenesis of motile primary cilia in the embryo (32). Kartagener's syndrome, a combination of defects that appear as abnormalities in nodal cilia, bronchial cilia and sperm flagella, is also caused by malfunctioning of the KIF3-dependent intracellular transport pathway. In anterograde transport pathways, KIF3 has been found in many typical neurons that lack cilia (16, 17, 35-37). In a specific subset of neurons in Drosophila, mutants lacking a KIF3 display reduced transport of the soluble enzyme choline acetyltransferase from cell bodies to the synapse (38).
Alzheimer's disease
Alzheimer's disease is primarily a neuronal disease. Previous studies have identified the possible role of amyloid precursor protein (APP) in the initiation or progression of Alzheimer's disease (39). APP is a transmembrane protein which in vivo is subjected to proteolytic cleavage (40). This cleavage produces the extracellular amyloid β-peptide of Alzheimer's disease and releases an intracellular tail fragment of unknown function. APP is synthesized in the endoplasmic reticulum, glycosylated in the Golgi apparatus, and then packaged into vesicular structures that are transported to the plasma membrane (41). Conventional kinesin (KIF5) was first discovered in a squid fast axoplasmic transport system, but its in vivo function and associated cargo have been uncertain for many years (1, 42). KIF5 is a heterotetramer consisting of two KHCs and two KLCs (43). KLCs consist of at least two structurally distinct regions: an N-terminal region of heptad repeats and a C-terminal region that has six-tetratrico peptide repeats (TRP) (44). Previous antisense experiments revealed that KIF5 was needed for APP transport in neurons (45). Co-immunoprecipitation, velocity gradient sedimentation and biochemical experiments confirmed this interaction and revealed that the cytoplasmic domain of APP binds directly to the TRP region of KLC (46, 47). In addition, analysis of KLC knockout mice revealed that APP transport in sciatic nerve axons strongly depends on KLC, showing dramatic reduction in its absence compared with normal animals. Based on the tight binding of APP to KLC, and the strong dependence of APP axonal transport on KLC, it was proposed that KIF5 is associated with a defined class of vesicular cargoes in the axon that contain APP and the transmembrane proteins involved in its proteolytic cleavage (47, 48). These findings suggest that axonal transport dysfunction or aberrant trafficking of APP might be an important element in the pathogenesis of Alzheimer's disease.
Diabetes
Glucose homeostasis in humans is dependent upon the catalytic activity of a glucose transporter protein, GLUT4 that responds to insulin by translocation from intracellular vesicles to the plasma membrane (49-51). Ablation of GLUT4 expression in either adipocytes or skeletal muscle of mice can lead to diabetes (52, 53). Insulin stimulates glucose uptake in muscle and adipose cells by mobilizing intracellular membrane vesicles containing GLUT4 glucose transporter protein to the plasma membrane. In the experiments of Czech's group, KIF5B is highly expressed in adipocytes and co-localizes with perinuclear GLUT4. Dominant-negative mutants of KLC blocked outward GLUT4 vesicle movements and translocation to the plasma membrane in response to insulin (54). These findings suggest that dysfunction of GLUT4 transport might be an important element in the pathogenesis of diabetes.
CONCLUSIONS AND OUTLOOK
This year is the eighteenth anniversary of the discovery of kinesin motor proteins, but the field has advanced far past the 'puberty' stage, and has reached a degree of maturity previously unforeseen. Since the first characterization of kinesin in 1985, many studies have revealed various members in this superfamily of proteins with correspondingly diverse functions. However, the relationship between kinesin and disease still raises a lot of intriguing questions. Understanding the interaction of kinesin and cargoes will be crucial in unveiling the intricacies of kinesin-related human disease and will further elucidate the mechanism of their regulation.


 XML Download
XML Download