

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The evaluation of hypertrophic cardiomyopathy (HCM) involves the careful exclusion of diseases that can cause increased left ventricular wall thickness, such as aortic stenosis, poorly controlled hypertension, and infiltrative processes such as cardiac amyloidosis and sarcoidosis. Fabry disease, an inherited deficiency of the enzyme α-galactosidase A, is a rare cause of left ventricular hypertrophy, especially, has drawn much attention for nearly curative treatments.1) Exogenous administration of the deficient enzyme has been shown to clear microvascular endothelial deposits, which are essential to disease manifestation, in the kidney, skin, and heart. In one study, this enzyme deficiency is found in 10% of male subjects with myocardial hypertrophy without definite cause.2) Whereas another study showed that approximately 6% of men and 12% of women with late-onset HCM actually had evidence of Fabry disease.3) If this storage disease is misdiagnosed as HCM, patients who could be treated effectively with medication might be subjected to aggressive procedures such as septal ablation or surgical septal myectomy. We report a case of Fabry disease which presented with HCM and tricuspid regurgitation and underwent an open heart surgery.
Case
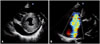
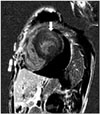
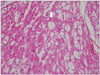
A 71-year-old female presented with dyspnea (New York Heart Association functional class III) and severe edema on both legs on July 2015. In 1999, she was diagnosed with non-obstructive HCM, and her symptom was stationary until January 2015 when her dyspnea and peripheral edema were aggravated and referred to our hospital. The initial blood pressure was 127/81 mm Hg. Routine hematologic tests showed iron deficiency anemia (hemoglobin: 9.7 g/dL) and normal coagulation profile. Chemical profile was within normal range including normal renal function (blood urea nitrogen/Cr: 11/0.84 mg/dL). Chest X-ray revealed a marked cardiomegaly (cardiothoracic ratio = 84%). A 12-lead electrocardiogram (ECG) showed atrial fibrillation with a mean ventricular rate of 56 bpm and ST-T change (Fig. 1). Echocardiography showed a markedly thickened wall of left ventricle and right ventricle with a high echo density and severe tricuspid regurgitation (Fig. 2). Cardiac magnetic resonance image showed a fuzzy delayed Gadolinium enhancement at the mid anterolateral and inferolateral myocardium of the left ventricle suggestive of infiltrative diseases such as amyloidosis and sarcoidosis (Fig. 3). For a confirmative diagnosis, endomyocardial biopsy was performed. Hematoxylin and eosin staining showed a diffuse vacuolization of myocytes and lateralization of nucleus (Fig. 4). Congo-red staining was negative and no definite sarcoid granuloma or amorphous amyloid deposition was identified, which excluded the possibility of cardiac sarcoidosis or amyloidosis. Therefore, initial pathologic diagnosis was vacuolar change of myocyte.
For the management of her symptom, cardiac surgery including tricuspid annuloplasty, Maze operation and right atrial reduction plasty was performed. After surgery, her symptom was improved to mild exertional dyspnea (New York Heart Association functional class I–II) and the edema on both legs decreased. ECG showed normal sinus rhythm. On echocardiography, tricuspid regurgitation was reduced to mild to moderate and a moderate resting pulmonary hypertension was noted with a maximal velocity of tricuspid regurgitation of 3.5 m/sec. During follow-up after cardiac surgery, a plasma α-galactosidase activity was checked for the screening of Fabry disease. The result of plasma α-galactosidase activity was around lower normal limit (30.1 nmol/hr/mg, reference range: 25-126 nmol/hr/mg). DNA analysis was implemented for confirmation and it revealed a heterozygote α-galactosidase mutation at exon 6 [c.901C>T (p.Arg301Ter)] (Fig. 5).
After the diagnosis, her history and physical findings were meticulously reviewed. Since teenager, she had pain on the palms and soles (acroparesthesia), hypohidrosis and a heat and cold intolerance. On physical examination, angiokeratomas were found on her lower abdomen. Ophthalmic examination showed a sign of corneal opacity and severe corneal verticillata (Fig. 6), and conjunctival vascular tortuosity. No retinal vascular tortuosity was found on fundoscopic examination. The pure tone audiometry showed a sensorineuronal hearing loss (Fig. 7). The albumin/creatinine ratio (141.4 mg/day, reference range: 0–100 mg/day) and the amount of 24-hour urine protein (7837.0 mg/g, reference range: 0–30 mg/g) were increased.
She started to be treated with enzyme replacement therapy (ERT) using α-galactosidase. On the family screening, her younger sister and her first son were turned out to be affected by Fabry disease. Her younger sister who was 66 years old had renal dysfunction. Her first son who was 44 years old had acroparesthesia on hands and feet and hypohidrosis since he was 9 years old. His proteinuria and abnormal ECG were detected since 2014, and left ventricular hypertrophy was observed on his echocardiographic examination.
Discussion
Fabry disease is a X-linked recessive metabolic disorder characterized by deficient activity of the lysosomal hydrolase, α-galactosidase A.4) This enzymatic defect leads to the progressive accumulation of glycosphingolipids, predominantly globotriaosylceramide (Gb3), within the lysosomes of endothelial cells. The clinical manifestations depend on the part of body which is damaged, and acroparesthesias, hypohidrosis, corneal opacities (cornea verticillata) and dysfunction of the kidney, brain, and heart can be developed.5)
Cardiac manifestation can be seen in almost all patients, but symptoms from the cardiovascular system appear much later than Gb3 accumulation starts. The mean age of cardiac symptom onset is 32 years and 40 years in male and female patients, respectively. The cardiovascular complications include myocardial hypertrophy of the left ventricle, thickening of the valves, dilatation of the ascending aorta and conduction disturbances. 6) In the end stage of cardiomyopathy caused by Fabry disease, myocardial fibrosis is extensive, and systolic and diastolic left ventricular functions become severely impaired.7) In our case, a severe ventricular hypertrophy was developed, which might resulted in atrial fibrillation and tricuspid regurgitation. Myocardial echo density was very high, resembling the typical feature of myocardial sparkling found in cardiac amyloidosis. However, ECG did not show a low voltage which is typically caused by interstitial infiltration of amyloid, and amyloid infiltration was not found in myocardial biopsy.
Fabry disease cardiomyopathy mimics HCM and infiltrative disease in respect to the morphologic and clinical features. Therefore, differential diagnosis is not easy and frequently relies on invasive studies such as endomyocardial biopsy or molecular and gene analysis. In particular, Fabry disease cardiomyopathy shares many clinical and morphologic features with HCM, and Fabry disease cannot be differentiated if we do not suspect and pay attention to systemic manifestations of this disease. However, some clues of Fabry disease can be found in heart. On echocardiography, patients with HCM show more frequently present with asymmetric septal hypertrophy and outflow tract pressure gradient in comparison with patients with Fabry disease. A binary appearance of left ventricular endocardial border, reflecting the endocardial and subendocardial compartmentalization of glycosphingolipid material, can be a good indicator of Fabry cardiomyopathy.8) Tissue Doppler imaging, and strain rate imaging can help to detect early diastolic dysfunction due to regional fibrosis in Fabry disease.9)10)
Cardiac magnetic resonance has no pathognomic finding of Fabry disease, but the difference of focal late enhancement pattern can be helpful. In most Fabry disease patients (92%), late gadolinium enhancement was noted in the basal infero-lateral walls.11) In Fabry cardiomyopathy, endomyocardial biopsy shows cardiomyocytes containing large perinuclear and cytoplasmic vacuoles representing accumulation of glycolipid material and the characteristic appearance of lamellar bodies inclusion in electron microscopy,12) whereas HCM shows only disarrayed hypertrophied myocytes without cytoplasmic vacuoles.
The diagnosis of Fabry disease starts from clinical suspicion. If there are 2 or more clinical problems among acroparesthesia or neuritic pain in hands or feet, persistent proteinuria of unknown cause, progressive renal impairment of obscure cause, unexplained HCM, angiokeratomas etc., high suspicion of Fabry disease should be raised, then galactosidase A activity assays should be checked for screening. In affected males, enzyme assays usually show severely diminished activity in plasma and leukocytes and Fabry disease diagnosis can be confirmed without further exam. However enzyme activity in heterozygote female is usually normal as in our case, confirmation exam such as detection of a gene mutation or tissue biopsy is needed, if the patient is suspicious of Fabry disease. After a Fabry disease-specific gene mutation is identified or cytoplasmic vacuoles in light microcopy or lamellar bodies inclusion in electron microscopy on biopsy tissue is noted, Fabry disease can be confirmed. All family members suspected of having Fabry disease should be tested, even if they have no symptoms. Patients with unexplained left ventricular hypertrophy should be checked if they have symptoms related to Fabry disease.
ERT with galactosidase is the specific therapy for Fabry disease. ERT stabilizes the levels of Gb3 in the endothelial cells of the myocardium leading to reduce fibrosis development, and consequently improves left ventricular function.13) However, the treatment is effective when the disease's state is early and myocardial fibrosis is in a low degree.14) Regarding other systems, treatment reduces neuropathic pain, stabilizes renal function and contributes to the regression of the major pathological complications as well as to an improvement in prognosis. 1)15)
Fabry disease might be easily undetected, and clinical suspicion is critical. Our case was not diagnosed with Fabry disease until cardiac surgery was performed to correct severe tricuspid regurgitation and atrial fibrillation. If Fabry disease was confirmed earlier and ERT was started, ventricular hypertrophy might be regressed before cardiac surgery was required.




 XML Download
XML Download