

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Despite recent advances in cardiopulmonary support strategies, dilated cardiomyopathy (DCMP) remains a life threatening disease in young patients.1)2) Early recognition and treatment of DCMP are crucial for good prognoses in this patient population.3)4) The clinical course of patients with DCMP that result in cardiogenic shock varies according to the etiology of their disease as well as patient age. Several cases of DCMP complicated by sleep apnea syndrome have reportedly been managed with continuous positive airway pressure.5) The volumetric expansion of the enlarged heart can compress adjacent structures in the thoracic cavity causing a number of related symptoms, especially in infants with soft cartilaginous bronchi.6)7) Therapeutic strategies for treating these issues vary according to the type of complication encountered.
Case
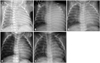
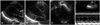
A corrected age 2-month-old female infant presented to our hospital with poor feeding and irritability for 7 days. The patient appeared pale and dusky on initial assessment. The infant was delivered prematurely at our hospital with a birth weight of 2.45 kg (50-75 percentile) and a height 47 cm (75 percentile) at 34+3 weeks of gestation (Fig. 1 A), and had an uneventful clinical course thereafter. There was no history of recent viral infection or family history of cardiac disease. The patient's body weight on admission was 5.1 kg (10-25 percentile) and height 60 cm (75-90 percentile). The patient exhibited signs and symptoms of shock with blood pressure 54 mmHg (in systole) and 31 mmHg (in diastole), heart rates 200 beats per minute, respiration rates 60 per minutes, body temperature 36.6℃. Chest roentgenogram showed unilateral haziness of the entire left lung and suspected cardiomegaly, while the right lung field appeared to be normal (Fig. 1 B). Echocardiography revealed a massively dilated left ventricle (LV) with poor contractility (Fig. 2 A). There was also evidence of severe atrioventricular valve regurgitation (Fig. 2 B), and the LV chamber appeared spherical in shape while the left atrium was not grossly enlarged (Fig. 2 C). The patient's LV ejection fraction (EF) was estimated to be 24.5%, with a diastolic LV inner diameter of 41.3 mm, end point septal separation of the anterior mitral valve of 17.3 mm (Fig. 2 D), and spherical index of 0.92 (reference; 0.60-0.73) (Table 1). No anatomical abnormalities of the coronary arteries were noted. In contrast to the markedly elevated N-terminal pro brain natriuretic peptide of > 35000 pg/mL and severe anemia with a hemoglobin of 6.5 g/dL, other cardiac enzymes including serum glutamic oxaloacetic transaminase, creatine phosphokinase, lactate dehydrogenase, creatine phosphokinase-MB, troponin-T, and C-reactive protein remained within normal limits throughout the patient's hospital stay (Table 2). Retrospective review of the chest roentgenogram taken at birth revealed equivocal cardiomegaly (Fig. 1 A), but no abnormal cardiopulmonary signs or symptoms were noted with respect to the previously recorded vital signs.
With aggressive cardiopulmonary support including inotropes (dopamine, dobutamine, and milrinone), diuretics, and continuous nitroglycerine (0.5 µg/kg/min) infusion with shock management, vital signs were relatively stable by the 3rd day of admission. Extracorporeal membrane oxygenator support or ventilator care were not applied throughout the entire course in intensive care unit. Studies performed to identify the etiology of DCMP including tests for enterovirus, adenovirus, cytomegalovirus, rubella, measles, Epstein-Barr virus, mumps, parvovirus, herpes virus, Varicella-Zoster, the hepatitis viruses, and cultures for several kinds of bacteria. These studies along with measured carnitine levels yielded non-specific findings. While LV systolic function improved gradually on echocardiography, the spherical contour and markedly increased inner diameter of the LV and valvular insufficiency showed little interval change prior to discharge. Intravenous medications such as inotropes and nitroglycerine were switched to oral medications including an angiotensin converting enzyme inhibitor, digoxin, and diuretics 10 days after admission as the patient was transferred to the general ward. On the 24th day of admission, the patient was discharged home in stable condition with a LVEF of 35%(Table 1).
Two months later at an outpatient follow-up visit, the patient continued to have decreased breath sounds with total haziness of the left lung field on roentgenography, making it difficult to assess the cardiac silhouette and size (Fig. 1 C). Chest computed tomography (CT) (Fig. 3A, B, and C) with 3 dimensional (3D) reconstruction of the airway revealed severe external compression of the left main stem bronchus between the enlarged left atrium and the aorta, resulting in atelectasis in the left lung (Fig. 3 D). As the patient did not show any signs of respiratory distress, she was treated with conservative management. The patient showed normal growth and development without any neurologic complications. With gradual improvement of cardiomegaly and LV function, the atelectasis resolved spontaneously within 2 months (Fig. 1 D and E). After a year of treatment with oral carvedilol, follow-up echocardiography showed normal LV systolic function and nearly normal LV contour (Fig. 4). One year from the onset of symptoms and signs, as an 14 month old female infants, the patient showed normal growth with body weight 11.4 kg (25-50 percentile), height 77 cm (25-50 percentile) and normal development without any neurologic complications. She is now managed at regular follow-up visits and is asymptomatic.
Discussion
It is often difficult to differentiate between DCMP and myocarditis. Since the patient in this case experienced sudden onset of massive cardiomegaly with fulminant heart failure, DCMP may have resulted from fulminant myocarditis secondary to a viral infection. Evidence of recent viral infection, however, could not be identified. DCMP typically causes dilatation of the mitral valve annulus, resulting in valve insufficiency and left atrial enlargement. Due to the intimate relationship between the airway and the left atrium, left pulmonary veins, and left pulmonary artery, if the mean pulmonary arterial pressure, mean left atrial pressure, and carinal angle increase, the likelihood of major airway compression is high.8) However, in this case the left atrium was not severely enlarged despite the nearly perfect spherical dilation of the LV chamber. In very young infants, soft cartilaginous main stem bronchi can be easily externally compressed. This results in atelectasis that is difficult to manage, and surgical intervention is sometimes required.9) In this case, the obstruction of the left main bronchus resolved completely without surgical intervention after 3 months of prolonged compression by the enlarged heart. Considering the various geometric changes that can occur with cardiomegaly, interventional strategies could potentially be tailored on a case by case basis with the help of cardiac CT imaging.10) We report a case of severe DCMP with massive cardiomegaly complicated by bronchial obstruction in which 3D CT was helpful for assessing detailed airway patency and choosing therapeutic strategies in an infant.




 XML Download
XML Download