

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Secundum atrial septal defect (ASD) is a common congenital heart disease identified in adulthood. Patients with ASD and left to right shunt are at risk for developing pulmonary arterial hypertension (PAH).1) However, a minority of patients (< 1%) develop precocious, severe pulmonary hypertension with shunt reversal.2) It is a current practice to assess the reversibility of pulmonary hypertension by means of pulmonary vasodilator testing during right heart catheterization in patients with shunt-related pulmonary hypertension.3) Patients with irreversible PAH are considered ineligible for shunt closure because of the risk of right ventricular (RV) decompensation after the intervention.4)
There are several reports about successful surgical or percutaneous ASD closure in patients with severe and seemingly irreversible pulmonary hypertension.4-6) Here, we report a patient who showed remarkable recovery of severe PAH after percutaneous closure following 1 year use of the endothelin receptor antagonist, bosentan (Tracleer, Actelion, Allschwil, Switzerland).
CASE
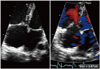
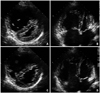
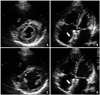
A 20-year-old woman presented with progressively worsening dyspnea for 6 months. She had no history of evaluation or treatment for her symptoms, although the symptoms started at the age of high school. Her symptoms became worse with time, and she presented with NYHA class III exertional dyspnea and orthopnea. Her height, weight, and body surface area were 156 cm, 49 kg, and 1.45 m2 respectively. Her vital signs were blood pressure 104/68 mmHg, heart rate 104/min, and respiratory rate 22/min. A regular heart beat was detected with wide fixed splitting of S2 at the pulmonary valve area, and no clubbing of the fingers or nails was observed. A simple chest X-ray showed cardiomegaly and a dilated pulmonary trunk with cephalization of pulmonary vascular marking. Electrocardiography revealed normal sinus rhythm with compatible findings of RV hypertrophy. A transthoracic echocardiogram revealed a large tissue defect of 29 mm with a bi-directional shunt through the interatrial septum (Fig. 1). The RV was prominently dilated and revealed significantly decreased contractility with a D-shaped left ventricle (LV) showing normal contractility (Fig. 2). There was no significant RV hypertrophy. The systolic pulmonary arterial pressure measured by maximal tricuspid regurgitation velocity (TR Vmax) was estimated at 95 mmHg (TR Vmax = 4.6 m/sec), which was roughly equal to systemic systolic blood pressure (Fig. 3). Her walking distance was 362 m on the 6-minute walking test with slightly decreased oxygen saturation from 98% to 92%. Further evaluations to identify the etiology of the pulmonary hypertension included a pulmonary function test, chest computed tomography angiogram and autoimmune panel, but all were negative.
Cardiac catheterization confirmed severe pulmonary hypertension with a mean pulmonary arterial pressure of 48 mmHg (76/36 mmHg absolute) and pulmonary vascular resistance (Rp) of 9.6 Wood units (WU) on room air. The mean systemic arterial pressure was 84 mmHg (97/73 mmHg absolute). After administering oxygen (10 L/min for 10 minutes via a nasal prong) and inhaling iloprost, her mean pulmonary arterial pressure was 50 and 46 mmHg respectively, and had not changed significantly. We failed to identify the pulmonary arterial responsibility after pulmonary vasodilator administration. Based on these findings, closure of the ASD was not performed. Instead, medical treatment including oral bosentan was started to improve patient's symptom.
Her symptoms and exercise tolerance improved from NYHA class III to NYHA class I to II after 1 year of using bosentan. She was able to walk 426 m during the 6-min walk test without desaturation. A follow-up echocardiogram showed no significant interval change except slight improvement in RV contractility compared to the initial echocardiogram, even though her symptoms were much improved. TR Vmax was 4.3 m/sec by continuous wave Doppler test, but there was no significant improvement in comparison with the initial test. Although the follow-up echocardiogram result was very disappointing, we decided to perform a cardiac catheterization and balloon occlusion test to confirm the change in Rp and possibly correct the ASD. We expected some favorable changes in pulmonary vascular physiology, because her exercise tolerance had much improved.
A follow-up cardiac catheterization revealed severe pulmonary hypertension with mean pulmonary arterial pressure of 45 mmHg (75/29 mmHg absolute). The mean aortic pressure was 74 mmHg (95/60 mmHg absolute). Although the patient still had severe pulmonary hypertension, Rp had decreased markedly from 9.6 to 4.2 WU and calculated pulmonary to systemic flow ratio (Qp/Qs) increased from 1.9 to 2.6 in comparison with the initial data. We performed the transient balloon occlusion test using a 34 mm sizing balloon for 10 minutes. As a result, mean pulmonary arterial pressure decreased to 36 mmHg (48/25 mmHg absolute), and mean aortic pressure increased to 85 mmHg (118/70 mmHg absolute). We decided to close the ASD with a transcatheter occluder device considering her young age and the high perioperative risk. We performed percutaneous closure of the defect with a 34 mm Amplatzer® septal occluder (AGA Medical Corp., Minneapolis, MN, USA).
No residual leak was observed on the follow-up transthoracic echocardiography. The patient was given antiplatelet therapy and was maintained on 62.5 mg bosentan bid with diuretics and an angiotensin receptor blocker for 1 year. Finally, her physical activity and symptoms were much improved as she could walk 554 m on the walk test 1 year after the device was deployed. A chest X-ray showed remarkably decreased pulmonary vascularity, but cardiomegaly remained. Follow-up transthoracic echocardiography showed increased left atrial size from 37 to 42 mm and LV chamber size of end-diastole/end-systole from 40/23 to 46/28 mm. RV size had decreased significantly, and RV contractility was much improved in comparison with the initial study, but a compressed LV still remained on the parasternal short axis view (Fig. 4). TR Vmax also decreased markedly from 4.6 m/sec to 3.0 m/sec (Fig. 2).
DISCUSSION
The presence of irreversible PAH in patients with ASD is still thought to preclude shunt closure. Closure of such shunts is associated with decrease of cardiac output and increase of right-sided heart failure and death. Thus, defect closure in these patients should be performed only if the benefits of abolishing the shunt outweigh the risks of surgical or percutaneous closure.7) An additional reason for concern when contemplating surgery in ASD patients with PAH is the high perioperative risk.
Perioperative risk can be reduced in patients with the defects amendable by percutaneous closure. Balint et al.8) from Canada reported the outcomes of percutaneous closure in patients with ASD and PAH. They concluded that transcatheter closure in patients with secundum ASD and PAH can be successfully performed in selected patient with good outcomes. However, they diagnosed pulmonary hypertension based on echocardiographic data and not cardiac catheterization data. Thus, they did not show the value of Rp and did not perform lung biopsies. Additionally, their patients were not so seriously compromised. Therefore, caution should be used when interpreting their results.
Several reports have shown good results of defect closure even in irreversible severe PAH with ASD after advanced therapy.4-6)9) Previously published articles suggested that pulmonary vasodilator therapy may offer patients with irreversible anatomical changes of the pulmonary vascular bed a change for further improvement of pulmonary pressures.7)10) In recent years, potent oral vasodilators aimed at the pulmonary circulation have been available, with promising results. The vasodilators appear to be effective in reducing Rp and symptoms in patients with near-systemic pulmonary pressure, previously thought to have irreversible pulmonary vascular disease.11)12) We thought that our patient was one of those who demonstrate a significant response to vasodilator therapy, because her physical activities and symptoms were much improved and mean pulmonary arterial pressure reduced from 45 mmHg to 36 mmHg with temporary balloon occlusion.
The exact mechanism of how preoperative pulmonary vasodilator therapy works on a pathologically irreversibly changed pulmonary artery is poorly understood. One of the possible hypotheses is that there is possible reverse remodeling of pulmonary vascular changes with endothelin receptor antagonists on the basis of their antiproliferative properties.12) Thus, even if there is no significant reduction of pulmonary arterial pressure after vasodilator therapy, pulmonary perfusion increases according to improved pulmonary vascular compliance. This result indicates that bosentan may increase pulmonary arterial perfusion by improving pulmonary arterial compliance rather than directly decreasing pulmonary arterial pressure. This makes it possible to decrease the calculated pulmonary arterial resistance and to close the ASD. Actually, in our case, the calculated Rp decreased remarkably from 9.6 to 4.2 and Qp/Qs increased from 1.9 to 2.6 after 1 year of bosentan, even though the pulmonary arterial pressure remained unchanged.
In summary, we experienced a case of a young woman who had ASD with irreversible severe pulmonary hypertension, but the pulmonary hypertension improved remarkably after successful percutaneous device closure following 1 year of bosentan.

 XML Download
XML Download