

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Despite the fact that up to 50% of heart failure patients have preserved left ventricular (LV) systolic function, treatment of diastolic heart failure remains largely empiric because there have been no large scale randomized trials evaluating the effects of specific pharmacological agents. Nonpharmacologic, pharmacologic and surgical approaches are available, although its efficacy has not been proven. Treatment should target the underlying pathological condition that causes the diastolic heart failure. For example, coronary artery disease, hypertensive heart disease, hypertrophic obstructive cardiomyopathy, and constrictive pericarditis provide relatively specific therapeutic targets, such as lowering of blood pressure, regression of hypertrophy and fibrosis, and treatment of ischemia, and a complete removal of constricting pericardium. Theoretically, pharmacological agents that facilitate myocardial relaxation and improve LV compliance would be ideal for the treatment of diastolic dysfunction. As a short-term therapeutic goal, manipulation of the loading conditions (preload, afterload) and control of the heart rate will be crucial. The treatment of underlying condition (e.g. hypertension) should be the ultimate goal for the treatment of LV diastolic dysfunction. Many of the drugs used for the treatment of symptoms of heart failure due to diastolic dysfunction are similar to those of systolic dysfunction, although the rationale for their use, the pathophysiological process that is being altered by the drug, and the dosing regimen may be entirely different. In this review, several therapeutic modalities including pharmacologic, nonpharmacologic, and surgical approaches for primary diastolic heart failure will be discussed.
Pharmacologic Therapy
Angiotensin converting enzyme (ACE) inhibitors and angiotensin II receptor blockers
ACE-inhibitors and angiotensin II receptor blockers show not only an effect on blood pressure but elicit a direct effect on the myocardium via the local renin-angiotensin system. These effects are essential for the regression of LV hypertrophy, and the improvement in the elastic properties of the myocardium.1)2) Several studies have documented that LV hypertrophy is more effectively reduced by ACE inhibitors than by other antihypertensive drugs,3)4) suggesting an effect on myocardial structure beyond that provided by the reduction of pressure overload. Experimental and clinical studies suggest that the interaction of angiotensin II with its type 1 (AT1) receptors plays a critical role in alterations of collagen type I metabolism and development of myocardial fibrosis in arterial hypertension. Myocardial fibrosis is responsible for an increase in intrinsic myocardial stiffness that may alter the LV diastolic properties. Chronic activation of the renin-angiotensin-aldosterone system has been shown to increase extracellular matrix fibrillar collagen and to be associated with increased stiffness. Brilla et al.3) randomized 35 patients with hypertension to receive either the ACE inhibitor lisinopril or the diuretic hydrochlorothiazide for 6 months. Only patients randomized to lisinopril had a significant reduction in myocardial collagen in association with improvement of diastolic dysfunction, although blood pressure reduction was similar in patients treated with either lisinopril or hydrochlorothiazide. López et al.4) studied 37 patients with hypertension randomly assigned to losartan or amlodipine for 12 months. Whereas myocardial collagen volume diminished significantly in losartan-treated patients, myocardial collagen volume remained unchanged in amlodipine-treated patients with a similar reduction of blood pressure. These findings suggest that the ability of antihypertensive drugs to decrease myocardial fibrosis is not exclusively linked to their capacity to reduce blood pressure but may also be related to their efficacy in interfering with non-hemodynamic fibrogenic factors (i.e. angiotensin II and TGF-1). Recently, it has been shown that losartan can induce a regression of myocardial fibrosis, which is associated with diminution of myocardial stiffness in patients with essential hypertension.5)
The recently reported Losartan Intervention For Endpoint reduction in hypertension study (LIFE)6) showed a reduction in cardiovascular end points with losartan when compared with atenolol in patients with hypertension and electrocardiographic evidence of LV hypertrophy. Similar results were reported from a direct comparative trial of angiotensin II receptor blockers and beta-blockers that despite similar reductions in blood pressure, regression of LV hypertrophy with irbesartan was greater than those attained with atenolol.7)
Elevated circulating levels of angiotensin II have been suggested as a mediator of exaggerated blood pressure response during exercise, which may contribute to the exacerbation of diastolic dysfunction. Warner et al.8) investigated the effects of angiotensin II blocker on exercise tolerance in patients with diastolic dysfunction and an exaggerated systolic blood pressure response during exercise. Although 2 weeks of therapy with losartan had no significant effect on diastolic function parameters such as isovolumic relaxation time, mitral E/A ratio, deceleration time of E wave, the hypertensive response to exercise was blunted with losartan, thereby increasing exercise tolerance and improving quality of life after 2 weeks of therapy. Confirmation of the favorable effects of ACE inhibitors on mortality has to be done in a larger patient population with diastolic dysfunction. CHARM-Preserved trial explores the effects of candesartan in patients who are older than 60 years and have diastolic dysfunction, LV ejection fraction ≥ 40%, and symptoms of heart failure.9) Median follow-up was 36.6 months. Although cardiovascular death did not differ between groups, fewer patients in the candesartan group than in the placebo group were admitted to hospital for heart failure once (230 vs. 279, p = 0.017) or multiple times. Thus, the finding from CHARM-Preserved study suggest that candesartan has a moderate impact in preventing admissions for heart failure among patients who have heart failure and LV ejection fraction higher than 40%. Recently reported I-PRESERVE trial was designed to study the effect of the angiotensin II receptor blocker irbesartan in patients with NYHA II-IV heart failure and an ejection fraction of 45% or more.10) A total of 4,128 patients aged 60 years or older were randomized to treatment with either irbesartan 300 mg per day or placebo. Over a mean follow-up period of just over 4 years, there was no difference in the incidence of the primary composite endpoint (all-cause mortality or CV hospitalization) between the treatment groups. Secondary endpoints and subgroup analyses also suggested no benefit of irbesartan over placebo in this patient group. The findings highlight the urgent need for a better understanding of the mechanisms underlying the syndrome of "heart failure with preserved ejection fraction", in order to identify appropriate treatments.
Diuretics
Diuretics are effective in reducing pulmonary congestion in some patients with diastolic heart failure, shifting the pressure-volume relation downwards. However, they must be used judiciously because the volume sensitivity of patients with diastolic dysfunction bears the risk that an excessive diuresis results in sudden drop of stroke volume.11) Therefore, diuretics should be used in patients with an evidence of fluid overload and initiated at low dose to avoid hypotension and fatigue due to decrease in stroke volume. Diuretics are most effective in patients with acute symptoms due to systemic hypertension or in patients with chronic heart failure who have evidences of fluid retention. Echocardiographic evaluation should be able to identify patients who benefit from diuresis and those in whom diuretics should not be used.
Beta-blockers
Beta blockers are thought potentially to improve diastolic filling indirectly by way of negative chronotropic effect and thus increase in the time for diastolic filling, although animal experimental data showed early diastolic relaxation is impaired by beta blockers, whereas it is enhanced by sympathetic stimulation.12) It has also been used for many years to control blood pressure and, thus, to reduce myocardial hypertrophy. The antihypertensive action of beta blockers with regression of LV hypertrophy seems to be also important for improve ment of diastolic filling. Due to favorable effects, such as reduction of blood pressure, regression of ventricular hypertrophy, increase of the ischemic threshold, these drugs can be used in diastolic heart failure, especially in the presence of hypertension or coronary artery disease and atrial or ventricular arrhythmias.13) Theoretically, it should be also beneficial in patients with exertional dyspnea by blunting heart rate response to exertion. However, their use in patients with advanced diastolic dysfunction (grade III or IV) must be done with great caution.
Ca2+-channel blockers
Although calcium channel blockers do not specifically improve diastolic function acutely,14)15) it has shown to improve diastolic filling during exercise in patients with heart failure and normal LV systolic function and impaired diastolic filling.16) A significant increase in exercise capacity and peak filling rate was observed after 5 weeks of therapy with verapamil as compared with placebo with no change in baseline systolic function and systolic blood pressure. Calcium channel blockers with negative chronotropic action such as verapamil or diltiazem may improve diastolic filling by the reduction in heart rate.17)18) Calcium blockers have been shown to reduce muscle mass in patients with hypertension which can be associated with an improvement in passive elastic properties of the myocardium.
Aldosterone antagonists
Aldosterone antagonists have been used in models of experimental hypertension for their effect on fibroblasts and cardiomyocytes growth.19) These experimental data have shown promising results with regard to passive elastic properties of the myocardium. The effect of spironolactone on morbidity and mortality in patients with severe heart failure due to systolic dysfunction has been investigated in a large clinical trial (RALES).20) A reduction in mortality of 27% has been reported in these patients but the specific effect of this drug on diastolic dysfunction is not clear, and may be due to the afterload reducing effect, changes in serum electrolytes (potassium sparing effect), reduction in LV mass or the antifibrotic action on the myocardium.21)22)
Nitric oxide donors
Nitric oxide (NO) is synthesized from the amino acid L-arginine by the actions of the enzyme NO synthase. In patients with dysfunctional endothelium, the loss of flow-mediated and catecholamine-stimulated endotheilum derived relaxing factor (EDRF) release allows the constrictor effects of catecholamines to act unopposed. Thus, the loss of EDRF may contribute to impaired dilator responses of epicardial and resistance vessel and thereby to myocardial ischemia, which slows ventricular relaxation and increases myocardial wall stiffness. Previous studies also indicate that diastolic function of the heart appears to benefit from exogenous NO whereas its endogenous production does not play a major role in myocardial relaxation.23)24) Similarly, NO donors have been shown to exert a relaxant effect on the myocardium which is associated with a decrease in LV enddiastolic pressure.25)
Digoxin and positive inotropic agents
Digitalis may produce an increase in systolic energy demands while adding to a relative calcium overload in diastole. These effects may not be clinically apparent under many circumstances, but during hemodynamic stress or ischemia, digitalis may promote or contribute to diastolic dysfunction.26) Although results of the Digitalis Investigation Group trial 26 suggested that patients with heart failure and a normal ejection fraction may have fewer symptoms and fewer hospitalizations if they are treated with digitalis, the utility of digitalis in the treatment of diastolic heart failure remains unclear, except in those with atrial fibrillation. When atrial fibrillation occurs, restoration of sinus rhythm should be attempted and is best achieved with electric cardioversion.
Although positive inotropic agents enhance the rate of LV relaxation, which could be beneficial in some patients with systolic dysfunction and diastolic dysfunction, it is generally agreed that positive inotropic agents are not beneficial for the treatment of isolated diastolic dysfunction.
Endothelin antagonists
Endothelin is an endothelium-derived peptide with potent vasoconstriction27) and mitogenic properties.28) Since increased levels of circulating and local endothelin have been reported in patients with heart failure, endothelin antagonsists may play an important role. In patients with congestive heart failure taking an ACE inhibitor, exercise tolerance was worst in patients with the highest endothelin-1 levels during exercise.29) A recent study investigating the role of angiotensin II and endothelin-1 on the response of LV diastolic function to exercise in an canine model of pacing-induced congestive heart failure showed that elevated angiotensin II and endothelin-1 contributed to the diastolic dysfunction in the resting state and a further increase in these neurohormonal levels during exercise led to further exacerbation of the diastolic dysfunction.30) Thus, blocking the effects of both angiotensin-II and endothelin-1 may be a more effective treatment strategy than blocking the action of either hormone alone in improving exercise tolerance in congestive heart failure.
Natriuretic peptide
The natriuretic peptides have beneficial properties in heart failure including preload reduction, vasodilation and renin suppression. Recent in vitro31) and in vivo32) studies demonstrated that brain natriuretic peptide (BNP) has a positive lusinotropic effect through their second messenger, cGMP. Clarkson et al.33) evaluated the effects of BNP on resting and exercise hemodynamics and neurohormones in 6 patients with isolated diastolic heart failure and observed that BNP infusion causes significant attenuation of the rise of pulmonary capillary wedge pressure and mean pulmonary artery pressure during exercise without effects on resting hemodynamic parameters. In response to BNP infusion during exercise, circulating BNP levels were significantly increased while circulating aldosterone was suppressed, as compared with the patients who were infused with a placebo. Beneficial hemodynamic and neurohormonal effects during exercise by BNP infusion demonstrated by Clarkson et al.33) suggest possible direct and indirect effects to improve operant diastolic function and potential therapy for isolated diastolic dysfunction.
HMG-CoA reductase inhibitors
3-hydroxy-3-methyglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins) have been shown to inhibit angiotensin II--mediated myocyte hypertrophy34)35) and to block intracellular signaling molecules implicated in cardiac hypertrophy.36-38) Recently, simvastatin, a pleiotropic HMG-CoA reductase inhibitor, has been shown to induce the regression of cardiac hypertrophy and fibrosis, and improved LV filling pressures in a transgenic rabbit model of human hypertrophic cardiomyopathy.39) The mechanism(s) by which simvastatin induces the regression of hypertrophy and fibrosis and improves cardiac function is likely to involve downregulating the levels of activated ERK1/2, the predominant stress-responsive intracellular signaling kinase involved in modulating cardiac hypertrophy.40) Simvastatin has also been shown to induce the regression of load-induced cardiac hypertrophy by reducing the activity of the angiotensin-converting enzyme and the cardiac content of angiotensin II.35)
General Measures
Exercise
Evidence from laboratory animals as well as human subjects suggests that long-term exercise training may improve diastolic function. Spurgeon et al.41) demonstrated in rats that 5 months of swimming exercise eliminated the age-dependent increase in dynamic stiffness coefficient. Other animal studies have shown increased rates of myocardial relaxation and improved diastolic function after exercise training,42)43) possibly mediated by enhanced calcium uptake by the sarcoplasmic reticulum44) or more efficient excitation-contraction coupling.43) Cross-sectional studies in humans have suggested that exercise training may improve diastolic filling in young subjects.45-48) It also has been shown that normal LV filling dynamics were observed in long distance runners with physiologic LV hypertrophy assessed by radionuclide, M-mode echocardiography, and pulsed Doppler echocardiography.49-51)
Regular aerobic exercise has a favorable impact on a number of coronary risk factors, such as smoking, hypertension, elevated plasma LDL cholesterol, reduced HDL cholesterol, elevated plasma triglycerides, obesity, diabetes mellitus, thrombogenic factors and postmenopausal status. Longitudinal studies that assessed cardiorespiratory fitness by exercise testing have almost unanimously shown an inverse relationship between fitness and risk of CAD in both men and women. Since prevention of myocardial ischemia is an important part of the treatment of diastolic dysfunction, regular exercise should be beneficial for the primary prevention of diastolic dysfunction.
Afterload control
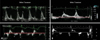
Hypertension is the most common cause of diastolic heart failure. Increase in systolic blood pressure results in an elevation of LV diastolic and mean left atrial pressures. Lowering of elevated blood pressure decrease the left atrial pressure and allows left ventricle to eject to a smaller end-systolic volume. In addition, normalization of systolic blood pressure enhances the myocardial relaxation and early filling. Myocardial ischemia associated with elevated systolic blood pressure may also be reduced or relieved. Uncontrolled hypertension promotes the development of LV hypertrophy. This hypertrophy impairs LV myocardial relaxation and distensibility contributing to diastolic dysfunction. Long-term therapy of hypertension results in regression of LV hypertrophy52) and it should improve diastolic performance of the left ventricle. In a recent study of 38 consecutive patients who had acute pulmonary edema and marked systolic hypertension, heart failure was due to exacerbation of diastolic dysfunction by hypertension, not by transient systolic dysfunction or mitral regurgitation.53) Although the control of hypertension is a key element in the treatment and prevention of diastolic dysfunction, satisfactory control of hypertension is achieved in relatively minor subset of this patient population.54-56) Fig. 1 is a Doppler tracing of transmitral flow and annular velocities obtained during the acute episode of pulmonary edema in a 66-year-old woman. The patient's initial blood pressure was 210/110 mmHg. The E/A ratio was 2 with deceleration time of 140 msec. The E/E' ratio was 37, indicating markedly elevated LV filling pressure (Fig. 1A). LV ejection fraction was 55%. Her subsequent diagnostic work up for hypertension revealed severe stenosis at the proximal left renal artery. Dilation with stenting of left renal artery was successfully performed. Follow-up Doppler echocardiography after normalization of blood pressure to 130/80 mmHg revealed improved mitral inflow filling pattern (from grade III to grade I diastolic dysfunction)(Fig. 1B) and the same LV ejection fraction. This case with acute pulmonary edema demonstrates that the dramatic acute effects of elevated blood pressures in causing diastolic dysfunction and diastolic heart failure are reversible with control of blood pressure.
Maintain sinus rhythm and optimal heart rate
Many patients with delayed relaxation and grade I diastolic dysfunction remain asymptomatic because of the compensatory augmentation of left atrial contraction. When atrial fibrillation develops in these patients, the resultant loss of atrial contraction will lead to elevation of the left atrial volume and pressure which in turn will precipitate symptoms of dyspnea at rest or with mild exertion. Hence it is crucial to maintain sinus rhythm in these patients with diastolic dysfunction. Maintaining optimal heart rate is also important, since tachycardia is poorly tolerated in patients with diastolic heart failure. Rapid heart rates cause an increase in myocardial oxygen demand and a decrease in coronary perfusion time, which can promote ischemic diastolic dysfunction even in the absence of epicardial coronary disease, especially in patients with LV hypertrophy. Second, a shortened diastole may cause incomplete relaxation between beats, resulting in an increase in diastolic pressure relative to volume. Third, hearts with diastolic dysfunction exhibit a flat or even negative relaxation velocity-versus-heart rate relationship, so that as heart rate increases, relaxation rate does not increase or may even decrease, which can then cause diastolic pressures to increase.57-59) Beta-blockers and some calcium channel blockers can thus be used to prevent excessive tachycardia and produce a relative bradycardia. Although the optimal heart rate must be individualized, an initial goal might be a resting heart rate of 60 to 70 bpm with a blunted exercise-induced increase in heart rate.60) By slowing heart rate, there will be more time for diastolic filling, which providing the ventricle with a prolonged diastolic time can allow adequate filling from a lower left atrial pressure in patients with diastolic dysfunction, especially in patients with grade I or II dysfunction. However, in patients with grade III or IV diastolic dysfunction, diastolic filling is nearly completed during early to mid diastole with little contribution from atrial contraction. These patients rely on a relatively fast heart rate to maintain adequate cardiac output. The drugs causing bradycardia in these patients with severe diastolic dysfunction (grade III or IV) may precipitate symptoms of low output state.
Prevention of myocardial ischemia
Marked changes in the diastolic properties of the left ventricle can occur in the presence of myocardial ischemia. The rate of ventricular relaxation is controlled primarily by the uptake of Ca2+ by the sarcoplasmic reticulum and by the efflux of Ca2+ from the myocyte. Because these Ca2+ movements are against concentration gradients, they are energyconsuming. Therefore, ischemia-induced ATP depletion interferes with these processes and slows myocardial relaxation. Successful treatment of ischemia improves diastolic relaxation and lowers ventricular diastolic and pulmonary venous pressures, thereby reducing dyspnea. Therefore, prevention of myocardial ischemia is an important part of the treatment of diastolic dysfunction.
Permanent Pacemaker
A variety of atrial, atrial-ventricular (AV), and interventricular and intraventricular conduction disturbances (IVCD) are often seen in the setting of chronic heart failure. These conduction disturbances produce suboptimal ventricular filling, a reduction in ventricular dP/dt, prolonged duration of mitral regurgitation, and ventricular dyssynchrony (seen as abnormal or paradoxical septal wall motion), which may further impair the pumping ability of the heart.61-64)
In the presence of a long AV delay and/or an IVCD, LV activation is delayed, but atrial activation is not. Hence, both early passive LV filling and the atrial kick may occur simultaneously, resulting in deceased total transmitral blood flow and diminished preloading of the left ventricle.65) These events are often seen as a fusion of the E and A waves on Doppler echocardiogram of transmitral blood flow. The adjusting the optimal AV interval can be seen by the return of normal E and A wave separation on Doppler echocardiogram of transmitral blood flow (Fig. 2). In addition, in the presence of a long PR interval and/or an IVCD, mitral valve closure may not be complete because atrial contraction is not followed by a properly timed ventricular systole. If the time lag is long enough, a ventriculoatrial pressure gradient may develop and cause diastolic mitral regurgitation.66) Since too short or too long P-R duration results in ineffective atrial contraction to ventricular filling65) maintaining optimal AV delay, defined as the shortest possible AV delay which allows complete ventricular filling and will optimize stroke volume and minimize diastolic mitral regurgitation.
A recent clinical study confirmed the therapeutic effects of cardiac resynchronization therapy by showing significant improvements in functional capacity, clinical status, and quality of life after cardiac resynchronization therapy.67) However, it is only applicable to a subset of patients with intraventricular conduction delay. No data are available so far regarding therapeutic efficacy of cardiac resynchronization in patients with diastolic dysfunction.
Surgical Treatment
Surgical treatment is helpful in subsets of patients associated with specific disease that causing diastolic dysfunction. As forementioned, myocardial ischemia can result in marked changes in the diastolic properties of the left ventricle. Coronary artery bypass graft surgery or percutaneous coronary intervention should be helpful for relieving myocardial ischemia and improving diastolic dysfunction in patients with ischemic heart disease.
Constrictive pericarditis is a form of diastolic heart failure as a fibrotic, thickened, and adherent pericardium restricts diastolic filling of the heart. The symmetrical constricting effect of the pericardium results in elevation and equilibrium of diastolic pressures in all four cardiac chambers. Surgical resection of the pericardium resulted in hemodynamic and symptomatic improvement in patients with constrictive pericarditis.
Hypertrophic obstructive cardiomyopathy is a genetic disorder associated with significant morbidity and mortality, including heart failure and sudden death. LV diastolic function is impaired in patients with hypertrophic obstructive cardiomyopathy due to impaired relaxation and increased chamber stiffness. In these patients, ventricular relaxation is related to systolic and diastolic hemodynamic loads and the extent of spatial and temporal heterogeneity of load and inactivation.68) Numerous studies have demonstrated the presence of nonuniform relaxation in patients with hypertrophic cardiomyopathy using several invasive and noninvasive techniques.69-71) Although regional fibrosis, hypertrophy, myocyte disarray, and ischemia can account for some of the regional heterogeneity, the presence of LV outflow tract obstruction results in a further imbalance of loads that can adversely affect regional function. Recently, nonsurgical septal reduction therapy has gained popularity as an alternative to surgery.72-74) LV hypertrophy assessed as wall thickness throughout the LV circumference was significantly reduced after nonsurgical septal reduction therapy. In addition, it has also been shown that improvement in LV relaxation and filling pressures 3 to 6 months to a year after nonsurgical septal reduction therapy.75)76)
Gene Therapy
Recent basic researches on molecular and genetic mechanism of diastolic dysfunction resulted in a newer therapeutic approach, such as gene therapy, in this specific disease entity. In mammalian hearts, aging is associated with impaired cardiac relaxation.77)78) Senescent myocytes are characterized by prolonged relaxation, diminished contraction velocity, a decrease in β-adrenergic response, and increased myocardial stiffness.79) This impairment in diastolic function contributes to the increased incidence of congestive heart failure in the elderly.80) A number of cellular and molecular mechanisms may contribute to the age-related defects. The abnormalities in cardiac relaxation have been attributed to a defect in SERCA2 activity.81-83) Schmidt et al.84) evaluated cardiac function in senescent rat using catheter-based adenoviral gene transfer to achieve global myocardial transduction of SERCA2. They found that overexpression of SERCA2 normalized both the maximal rate of decline of LV systolic pressure and the LV time constant of isovolumic relaxation. These data demonstrate the feasibility of achieving important functional cardiac effects through in vivo somatic gene transfer in a rodent model of senescence and it also demonstrated that the overexpression of SERCA2 through adenoviral gene transfer in senescent rat hearts improves diastolic function and restores contractile reserve. Thus, targeting SERCA2 may be an important strategy to improve diastolic function in the aging myocardium.
Recently, it has been shown that the decay phase of the calcium transient is accelerated by parvalbumin expression at the level of the isolated adult cardiac myocytes in vitro.85) Parvalbumin is a soluble, small-molecular weight intracellular calcium-binding protein that is highly expressed in ultrafast contracting/relaxing striated muscle fibers, but is not naturally expressed in the heart.86) Parvalbumin functions as a delayed calcium sink in fast muscle based on the relative affinities of its two calcium/magnesium binding sites.87)88) Parvalbumin is therefore ideally designed to speed the rate of decline in intracellular calcium, with additional advantages of this occurring via a non--ATP-dependent process. Although parvalbumin is not naturally expressed in the heart, Szatkowski et al.89) have shown that parvalbumin gene transfer to the heart in vivo produces levels of parvalbumin characteristic of fast skeletal muscles, causes a physiologically relevant acceleration of heart relaxation performance in normal hearts, and enhances relaxation performance in an animal model of slowed cardiac muscle relaxation. They suggested that parvalbumin may offer the unique potential to correct defective relaxation in energetically compromised failing hearts because the relaxation-enhancement effect of parvalbumin arises from an ATP-independent mechanism.
Therapeutic Approach Based on Symptoms and Doppler Echocardiographic Findings
Beta blockers, calcium channel blockers, and ACE inhibitors are the most frequently used agents for treating diastolic heart failure. Diuretics are appropriate therapy for the relief of congestion and edema. However, caution should be taken to avoid overdiuresis, which may results in intravascular volume depletion and resultant hypotension or prerenal azotemia. Based on the patterns of various diastolic parameters, diastolic dysfunction can be graded as follows: grade 1, impaired relaxation; grade 2, pseudonormalized pattern (impaired relaxation with moderately elevated mean LA and LV filling pressures); grade 3, reversible restrictive pattern (impaired relaxation with markedly elevated mean LA and LV filling pressures); grade 4, irreversible restrictive pattern.90)91)
In patients with grade 1 diastolic dysfunction, main symptom is exertional dyspnea. Many elderly subjects and patients with hypertension or LV hypertrophy have Doppler echocardiographic evidence of impaired diastolic function, but do not have any symptoms of heart failure at rest. In patients with a similar diastolic function at rest, there will be a wide spectrum of alterations in diastolic function during exercise. In this setting, it is important to assess the diastolic function during stress or exercise with provocation of symptoms. Revealing the deterioration of LV filling pattern during exercise will facilitate the stratification of diastolic functional abnormality. In general, excessive tachycardia should be avoided. ACE inhibitors or angiotensin II receptor blockers are probably helpful to reduce the progression of the diastolic disease in this stage.
In patients with grade 2 or 3 diastolic dysfunction (abnormal relaxation and elevated filling pressures), the addition of diuretics should be considered due to elevated filling pressures. Adequate neurohormonal modulation by ACE inhibitors or angiotensin II receptor blockers should be continued to prevent further progression of the disease process. A restrictive filling pattern (grade 3 diastolic dysfunction) is associated with worse functional class, reduced exercise tolerance and a worst prognosis. Since most of the filling of the left ventricle occurs in early diastole, prolonging the diastolic filling time with a beta blocker would not be beneficial or may be harmful due to decrease in cardiac output in accordance with decrease in heart rate. Increase in diuretics may be needed to reduce the LV preload.
In patients with grade 4 diastolic dysfunction, adjunctive therapy should be considered and non-pharmacological therapy, such as cardiac transplantation, may be considered. For this decision, frequent reassessment of cardiac function, including systolic and diastolic function, should be performed (Fig. 3).
Future Directions
Diastolic dysfunction represents a growing clinical challenge in need of novel therapeutic approaches. Improvements of our understanding of the molecular pathogenesis of diastolic dysfunction and its incorporation into the diagnostic and therapeutic approaches will enhance the patient management in this disease population. Novel approaches will undoubtedly improve our evaluation and treatment of this entity in future. This makes it even more important to assess diastolic function and its functional reserve.
Despite the high prevalence, substantial morbidity, and significant mortality of diastolic heart failure, there has been few prospective, randomized, blinded pharmacologic trial data to guide clinical decisions. Fortunately, large outcome studies have been presented recently but the findings highlight the urgent need for a better understanding of the mechanisms underlying the syndrome of 'heart failure' with preserved ejection fraction or diastolic heart failure, in order to identify appropriate treatments. With the results of further future trials as well as better diagnostic test in the future, physicians should be in a better position not only to diagnose diastolic dysfunction or heart failure but also to manage it more effectively.


 XML Download
XML Download