

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Ischemic heart disease (IHD) secondary to acute myocardial infarction is among the most prevalent health problems in the world and is a major cause of morbidity and mortality. In view of this, a major research effort in cardiovascular medicine has been to develop approaches to salvage the myocardium at risk of necrosis in patients with acute myocardial infarction (AMI). After an AMI, early and successful myocardial reperfusion with the use of pharmacological thrombolytic agents or mechanical revascularization, i.e., primary percutaneous coronary intervention (PCI), is the most effective strategy for reducing the damage to the myocardium and improving clinical outcome. However, reperfusion of the ischemic myocardium can induce some injury. This phenomenon, termed myocardial reperfusion injury, can blunt the beneficial effects of myocardial reperfusion. Reperfusion contributes to lethal injury following prolonged periods of ischemia (Fig. 1).
Myocardial reperfusion injury was first postulated in 1960 by Jennings et al.2 as significant morphological alterations appearing after the onset of reperfusion, including cell swelling, contracture of myofibrils, disruption of the sarcolemma, and the appearance of intramitochondrial calcium phosphate particles. The very existence of lethal reperfusion injury was vigorously debated3 by those who maintained that lethal injury was expressed or hastened by reperfusion on the one hand4 and those who advocated reperfusion as a contributor to de novo injury.5
Mitochondria make up 30% of the volume of a single cardiomyocyte, underlining the importance of their traditional role as the ATP producing 'powerhouse' of the cell. However, it is clear that this complex organelle plays a variety of roles within the cardiomyocyte that extend beyond its established function as the cellular powerhouse. In this regard, the most significant discovery was its critical role as an arbitrator of cell death6 in addition to its function in cellular survival.
Administration of nitric oxide (NO) or NO donors prior to ischemia attenuates the consequences of myocardial ischemia-reperfusion (IR); that is, it reduces infarct size and endothelial dysfunction.7 NO synthesized by essentially all cardiac cell types is an ubiquitous cellular messenger that plays a key role in regulating cardiac function.8-12 NO is a highly diffusible gas that spreads very rapidly from its site of synthesis and is a free radical highly reactive with other species, notably oxygen, superoxide, and iron-containing heme groups that act as NO scavengers. For this reason, the half-life of NO is limited to seconds and its effects are localized to where it is synthesized. NO generated within the cardiomyocytes can exert intracrine effects or modify the functional properties of adjacent cardiomyocytes. NO generated from noncardiomyocyte sources (coronary, endocardial, and endothelial cells; autonomic nerves and ganglia; and blood-formed elements) can exert direct effects on cardiomyocytes and indirect effects by modulating coronary blood flow or autonomic transmission.8-12 The heart produces NO on a beat-to-beat basis in response to changes in coronary flow and myocardial loading. In rabbit hearts, NO levels reach peak values during diastole (~2.7 mM in the endocardium and ~0.93 µM in the myocardium) and are lowest during systole (0.67 and 0.26 µM, respectively); NO concentrations are 15% lower in rat hearts.13
Molecular Events during Lethal Reperfusion Injury
1. Oxidative stress
Although low levels of oxygen radicals and oxidants are normally formed in cells and play important roles in cellular homeostasis, mitosis, differentiation, and signaling,14 following ischemia and reperfusion, radical formation is greatly increased, thus triggering cellular injury. Although mammalian cells including cardiomyocytes express endogenous free radical scavenging enzymes,15 such as superoxide dismutase (SOD), catalase, and glutathione peroxidase, these antioxidative defenses are overwhelmed after ischemia and reperfusion. The oxygen radical hypothesis is of particular importance because it potentially can explain each of the other mechanisms of reperfusion injury. It has been demonstrated that exogenous free radicals cause cellular calcium loading with inhibition of the sarcoplasmic reticulum calcium ATPase and inhibition of the sodium potassium ATPase leading to sodium-mediated calcium gain.16 Oxygen radicals cause lipid peroxidation that can result in cell membrane breakdown causing cell swelling. It has been suggested that oxygen radicals result in the chemotaxis of neutrophils that in turn can lead to white cell plugging of capillaries and microvascular compression. In addition, white cells that are chemotaxed and activated are potent sources of further oxygen radical generation.17 Oxidative stress during myocardial reperfusion also reduces the bioavailability of the intracellular signaling molecule, nitric oxide, thereby removing its cardioprotective effects. These effects include the inhibition of neutrophil accumulation, inactivation of superoxide radicals, and improvement of coronary blood flow.18
2. Calcium paradox
At the time of myocardial reperfusion, there is an abrupt increase in intracellular Ca2+ that is secondary to sarcolemmal-membrane damage and oxidative stress-induced dysfunction of the sarcoplasmic reticulum. These two forms of injury overwhelm the normal mechanisms that regulate Ca2+ in the cardiomyocyte; this phenomenon is termed the calcium paradox.
The result is intracellular and mitochondrial Ca2+ overload, and this excess of Ca2+ induces cardiomyocyte death by causing hypercontracture of the heart cells and mitochondrial permeability transition pore (PTP) opening.19
3. pH paradox
The rapid restoration of physiologic pH during myocardial reperfusion, which follows the washout of lactic acid and the activation of the sodium-hydrogen exchanger and the sodium-bicarbonate symporter, contributes to lethal reperfusion injury. This phenomenon is termed the pH paradox.20 In neonatal rat cardiomyocytes, experimental studies have shown that reoxygenation with acidic buffer is cardioprotective;21 this effect may be mediated by the inhibition of mitochondrial PTP opening.22
4. Inflammation
After an acute myocardial infarction, the release of chemoattractants draws neutrophils into the infarct zone during the first 6 hours of myocardial reperfusion, and during the next 24 hours they migrate into the myocardial tissue. This process is facilitated by cell-adhesion molecules. These neutrophils cause vascular plugging and release degradative enzymes and reactive oxygen species.23
5. Metabolic modulation
Several experimental and numerous clinical studies have examined the cardioprotective potential of therapy with glucose, insulin, and potassium administered as an adjunct to myocardial reperfusion.24,25 These studies have been conducted on the premise that ischemic myocardium benefits more from metabolizing glucose than from fatty acids.26
A recent very large randomized controlled study from several centers reported no cardioprotective benefit from therapy with glucose, insulin, and potassium (GIK) as an adjunct to myocardial reperfusion in patients with acute myocardial infarction.27 The delay in initiating this therapy, the prolonged period of myocardial ischemia, and high and potentially damaging glucose levels have all been cited as reasons for the lack of cardioprotection. As alternatives to GIK solution, exenatide, an analog to glucagon-like peptide 1, reduces infarct size and improves cardiac function in a porcine model of ischemia and reperfusion injury.28
6. Mitochondrial K+ATP channel
One of the most important mitochondrial proteins recognized to be involved in preconditioning is the K+ATP channel. The actual mechanism by which K+ATP channel opening elicits cardioprotection is not yet clear, and debate surrounds the order of events with respect to reactive oxygen species (ROS) generation (i.e., evidence suggests both that ROS generation is an upstream trigger for K+ATP channel opening and that K+ATP channel opening is a trigger for ROS generation).29 Nevertheless, from the standpoint of NO signaling, several studies have shown that NO can directly affect the K+ATP channel via S-nitrosation.30 In addition, the mito-K+ATP channel is thought to be a downstream target for PKG in classic cGMP/NO signaling.31 However, in the latter case, the mechanism by which PKG signaling is transmitted into the mitochondrion remains unclear.32 Furthermore, all studies on the K+ATP channel are subject to several caveats. First, much controversy surrounds the actual existence of bona fide K+ATP channel subunits (KIR, SUR) in mitochondria, with conflicting results surrounding the use of antibodies and Western blots.33,34 Second, many of the pharmacologic tools used to probe K+ATP channel function are nonspecific. Most notably, the K+ATP channel opener diazoxide is a known inhibitor of complex II,35 and a protonophoric uncoupler,36 whereas the K+ATP channel 5-HD is a β-oxidation substrate.37 The non-K+ATP channel effects of both diazoxide and 5-HD were recently reviewed extensively,37-39 with the result that a large proportion of the literature on this topic may require reassessment. The mechanism of K+ATP-mediated protection is thought to include mild mitochondrial swelling, which may affect the formation of the PTP.40 Although it has been argued that the small K+ fluxes that would result from opening of K+ATP channels (and K+Ca channels) in ischemic preconditioning (IPC) would mildly uncouple mitochondria, it should be recognized that the magnitude of such K+ fluxes is not large enough to account for the magnitude of H+ leak seen in IPC.41 In a human study, sildenafil was shown to prevent endothelial dysfunction induced by ischemia and reperfusion via opening of ATP-sensitive potassium channels, and the proposed mechanism of the effect of sildenafil was direct and cGMP-mediated opening of K-ATP channels.42
7. Mitochondrial permeability transition pore
The mitochondrial PTP is a nonselective channel of the inner mitochondrial membrane. Opening the channel collapses the mitochondrial membrane potential and uncouples oxidative phosphorylation, resulting in ATP depletion and cell death.43 During myocardial ischemia, the mitochondrial PTP remains closed, only to open within the first few minutes after myocardial reperfusion in response to mitochondrial Ca2+ overload, oxidative stress, restoration of a physiologic pH, and ATP depletion.22,44 The overproduction of ROS and mitochondrial Ca2+ overload are known to trigger PTP formation. Downstream effects as a result of PTP opening include cytochrome c release, mitochondrial swelling, membrane depolarization, inhibition of ATP production, caspase activation, apoptosis or necrosis or both, and additional ROS production, which initiates mitochondrial and cellular dysfunction.
At pathologically high levels, NO can induce apoptosis both via the activation of p53, and via release of cytochrome c from the mitochondrial inner membrane and activation of caspases.45-47 Although high levels of NO can cause PTP formation, smaller concentrations of NO inhibit PTP formation.48 Lower levels of NO protect the mitochondrion against apoptotic agents such as TNF-α, serum starvation, hypoxia, and H2O2.49,50
Mechanisms by which NO inhibits PTP formation include the prevention of Bcl-2 cleavage by caspase 3,45 mild dissipation of the mitochondrial membrane potential, and inhibition of Ca2+ uptake.48 In terms of cardioprotection, the NO donor DETA-NONOate was found to inhibit cyclosporin A-sensitive Ca2+-induced mitochondrial swelling in mitochondria isolated from hearts subjected to IR.48,51 Another study observed that aged endothelium, with decreased eNOS activity, and eNOS knockout mice were more susceptible to proapoptotic stimuli, which were reversed by NO donors.52 Therefore, the mitochondrial PTP is a critical determinant of lethal reperfusion injury, and as such it is an important new target for cardioprotection.
Nitric Oxide: Acting on Mitochondria for Cardioprotection against Ischemia-Reperfusion Injury
1. Mitochondrial dysfunction in IR injury
The hallmarks of cardiac IR injury are found to occur on reperfusion of the myocardium. In terms of the mitochondria, reperfusion injury affects the oxidative phosphorylation (Ox-Phos) pathway, including the ETC,53-55 adenine nucleotide translocase (ANT),56,57 and Krebs cycle58,59 enzymes. In addition to Ox-Phos, reperfusion injury also leads to cardiolipin oxidation,55,60,61 the induction of a large proton leak across the mitochondrial inner membrane,41,62 Ca2+ overload, overproduction of ROS, PTP opening, and cell death.62-73 A majority of these observations have been made in mitochondria isolated from hearts after reperfusion, meaning that the time course of mitochondrial damage during reperfusion injury is difficult to study (because mitochondrial isolation typically takes 1~2 h). The exact timing of mitochondrial damage during IR is an ongoing subject of investigation.
We demonstrated that MI might be the cause of mitochondria swelling and the changes in adhesion force and stiffness of mitochondria by use of a novel technique: atomic force microscopy (AFM).73,74 Mitochondrial changes such as swelling and irregularity of shape were also found in the infracted area in IR rat heart (unpublished data). Contrary to our findings in the AFM study, Brady et al.75 were the first to demonstrate extensive fragmentation of mitochondria in HL-1 cells (a murine atrial-derived cardiac cell line) in response to a sustained episode of simulated ischemia, changes that persisted into simulated reperfusion. Interestingly, in that study, the authors observed that reperfusing the ischemic cells with SB203580, a pharmacological p38MAPK inhibitor, caused the mitochondria to refuse and regain their elongated structures once more, which suggests that p38MAPK activity during reperfusion may have contributed to the detrimental changes in mitochondrial morphology.75 The authors have gone on to demonstrate in HL-1 cells that ischemia-induced mitochondrial fragmentation is associated with the translocation of Drp1 from the cytosol to the mitochondria (a process that is required for its pro-fission activity) and that the fragmentation process can be largely prevented by transfection with Drp1K38A (the dominant-negative construct of Drp1).76 Furthermore, in the adult murine heart, we demonstrated the fragmentation of IF cardiac mitochondria in the EM study. Plotnikov et al.77 demonstrated that pretreatment with an antioxidant could prevent ischemiainduced mitochondrial fragmentation. The mechanisms underlying mitochondrial fragmentation during ischemia are currently unclear, although we speculate that cytosolic calcium overload and ROS may be contributory factors. One can speculate that by interfering with cellular respiration, mitochondrial fragmentation may result in the production of ROS. In this respect, using primary renotubular cells, Plotnikov et al. demonstrated that pretreatment with an antioxidant could prevent ischemia-induced mitochondrial fragmentation.77 Interestingly, in that particular study, insulin was also reported to prevent ischemia-induced mitochondrial fragmentation, although the mechanism was not further investigated. In the context of diabetes, mitochondrial fragmentation induced by hyperglycemia has also been reported to result in the production of ROS; however, the actual interplay between mitochondrial fragmentation, ROS production, and mitochondrial respiration requires further investigation.
Despite evidence for mitochondrial dysfunction occurring on reperfusion, the degree of dysfunction also depends on the length of the ischemic insult, suggesting that ischemia itself is detrimental. Accumulation of Ca2+ and ROS generation does occur within the mitochondrion during ischemia, despite the mitochondria being deenergized.78 These events are linked to ischemic hypercontracture and the reversal of mitochondrial ATP synthase to use glycolytic ATP to maintain membrane potential. Thus, a reconciling paradigm is that the degree of mitochondrial dysfunction during ischemia, which is a function of the length of ischemia, is a harbinger of more severe mitochondrial dysfunction on reperfusion. In other words, more severe dysregulation of the mitochondria and contractile machinery during ischemia (as indicated by hypercontracture) leads to more severe pathology on reperfusion.
2. Mitochondrial role in cardioprotection
In contrast to the detrimental effects of prolonged ischemia, brief periods of ischemia initiate cardioprotective signaling cascades that preserve both myocardial and mitochondrial function during subsequent prolonged ischemia. This endogenous cardioprotective event was discovered more than 2 decades ago and is known as ischemic preconditioning, or IPC.79 Two "windows" of protection are elicited by IPC: the first acute phase is triggered within minutes and lasts 2 to 3 h, and a later delayed protective phase takes ~24 h to develop and lasts up to 72 h.
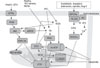
Fig. 2 shows several of the signaling pathways implicated in IPC. Several well-known and emergent therapeutics also trigger the same pathways, including pharmacologic preconditioning (PPC) agents [e.g., opioids, adenosine mimetics],80 anesthetic preconditioning (APC) with volatile anesthetics,81 and physical interventions such as slow or intermittent reperfusion (ischemic postconditioning).82 Thus, x-PC signaling continues to be an area of interest, not only because it can help to explain how these therapeutic molecules work, but also because it provides a deeper understanding of preconditioning signaling pathways, which offers the hope of improved therapeutics to be developed in the future.
While the spectrum of available cardioprotective strategies continues to grow, one concept that emerges from Fig. 4 is that the mitochondria are the downstream targets of most if not all of x-PC signaling. Key mitochondrial events include mild uncoupling (H+ leak), opening of mitochondrial K+ATP channels, inhibition of mitochondrial Ca2+ overload, attenuation of ROS generation, and inhibition of PTP opening at reperfusion.83-86 Notably, several of the mitochondrial events associated with IR injury are thought to occur in a limited manner during IPC. For example, a somewhat paradoxical situation is proposed wherein transient "flickering" of the PTP during the triggering or initiation phase of IPC is thought subsequently to prevent large-scale PTP opening during IR injury.87 In addition, limited ROS generation has been found to be essential in IPC signaling cascades, and the cardioprotective response was found to be blocked by antioxidants.88 Similar to PTP opening and ROS production, a large increase in H+ leak is found to occur in IR injury (affecting mitochondrial ATP synthetic capacity). However, a small reversible increase in H+ leak is seen during IPC, which may act to diminish ROS generation and Ca2+ overload at reperfusion.89 Pharmacologic preconditioning agents, such as diazoxide (a K+ATP channel agonist) and dinitrophenol (DNP, which induces mitochondrial proton leak), also elicit cardioprotection in IR injury by attenuating mitochondrial PTP formation, ROS generation, and H+ leak.87,90,91
The overarching principle of x-PC signaling can be summarized in the proverb "what doesn't kill you makes you stronger." It is therefore important to note that this principle is played out in its entirety in the microcosm of the mitochondrion. Fig. 2 highlights a multitude of upstream signaling pathways converging on the mitochondria to elicit cardioprotection.
3. The divergent roles of NO -IR injury
When considering the roles of NO in any cell-signaling process, it is important to discuss the sources of NO generation. Nitric oxide synthase (NOS) enzymes produce NO endogenously and are regulated by many of the upstream signaling pathways that are activated during IR and x-PC (Fig. 4).92 Studies on the cardioprotective and deleterious effects of NO have involved manipulation of endothelial NOS (eNOS) and inducible NOS (iNOS) activity. Attenuation of myocardial infarct size and preservation of left ventricular developed pressure and lower left ventricular end-diastolic pressure have been observed in mice overexpressing eNOS in a variety of models of ischemia-alone or IR injury, when compared with wild-type mice.93-95 The NOS-dependent protection was found to be abrogated with the administration of a commonly used NOS inhibitor, N(G)-nitro-L-arginine methyl ester (L-NAME).93 In agreement, studies using eNOS knockout mice have found an augmentation in infarct size96 and myocardial necrosis97 after IR injury. In terms of timing, eNOS is activated during the early window of preconditioning and activates transcription factors and other enzymes, including iNOS, in the late window of protection.98 The activation of eNOS has also been reported to contribute to the cardioprotective effects of postconditioning (brief intermittent reperfusion periods after ischemia). As seen in the eNOS transgenic and knockout studies, the cardioprotective effect of postconditioning was sensitive to L-NAME.98,99 Furthermore, support for the essential role NOS plays in cardioprotection is seen with the sexual dimorphism in response to IR injury; estrogen has been found to increase NOS expression, which may help to explain why premenopausal women have a lower risk of heart disease than do age-matched men.100
Several studies suggest the presence of a mitochondrial isoform of NOS (mtNOS), although the reproducibility of these studies appears to be limited to a small number of laboratories.101-103 Several recent articles have questioned the existence of mtNOS, with major controversies surrounding the purity of mitochondria, a 150,000-fold variation in the reported rates of NO generation, and several experimental artifacts in NO-measurement systems.104,105 It was reported that a NOS protein in the plant Arabidopsis thaliana (atNOS) is targeted to the mitochondrion, and more recently, a mammalian orthologue of this protein was proposed as a candidate for mtNOS.106,107 However, further investigation has found that the protein is a GTPase, not a NOS.108-110 Thus, the search for a unique mtNOS protein continues.
NOS enzymes have also been shown to play a deleterious role in IR injury. For example, iNOS-/- mice were shown to have lower mortality and enhanced left ventricular contractility when compared with wild-type mice after coronary occlusion.111 Also, exposing mitochondria to high concentrations of NO (micromolar) has been shown to initiate PTP opening.48 These results, along with other studies, have defined NO as a dual-faced molecule in IR injury, which contributes to both cardioprotective and deleterious signaling pathways within the myocardium. In this regard, understanding how to deliver NO (i.e., timing, concentration, location) may facilitate beneficial therapeutic exploitation of NO signaling in IR injury, while minimizing the deleterious effects of NO.
4. NO and mitochondria-dependent cardioprotection
Increasing evidence indicates that NO is a key mitochondrial regulator; for a number of reasons, the mitochondrion can be considered a cellular "hub" for NO signaling. First, mitochondria within the cardiomyocyte are in close proximity (1~2 µm) to the production site of NO.112,113 Second, NO is freely diffusible and partitions into membranous environments such as mitochondria, which contribute ~30% of the typical cardiomyocyte volume.114 Last, mitochondria are enriched in metal centers and thiols, and they generate ROS that interact with NO to produce several secondary intermediates important for NO signaling. The detailed NO-dependent modification of biomolecules is beyond the scope of this review and is not described further.
Before describing the protective effects of NO signaling, it is important to briefly discuss the deleterious side of NO at the mitochondrial level. Many of the deleterious effects can be attributed to the overproduction of NO by iNOS or the administration of NO donors at high concentrations, which causes irreversible oxidation of proteins, lipids, and DNA. Indeed, well before the identity of endothelium-derived relaxing factor was known, it was discovered that macrophages were able to generate a species (now identified as NO) that could inhibit cellular respiration.115 In contrast, lower endogenous production of NO and smaller concentrations of NO donors have been found to protect mitochondria during situations such as IR injury. An example of this is the dose-response dependence of isolated mitochondria to PTP opening with NO treatments.48 High levels of NO (>5 µM) were found to induce PTP formation, whereas lower levels of NO (100 nM to 1 µM) were found to inhibit pore opening. The following effects of NO on mitochondrial function are defined predominantly in terms of cardioprotection, because they are compared with x-PC agents that induce NOindependent cardioprotection.
Summary
For patients presenting with an AMI, early and successful myocardial reperfusion by means of thrombolytic therapy or primary PCI is the most effective interventional strategy for reducing infarct size and improving clinical outcomes. The process of myocardial reperfusion itself, however, can induce injury to the myocardium, thereby reducing the beneficial effects of myocardial reperfusion. Cardiomyocyte death is associated with the irreversible, lethal form of myocardial reperfusion injury. For this reason, lethal reperfusion injury would be expected to adversely affect clinical outcomes after an AMI, and it may contribute to mortality despite early and successful reperfusion.
NO and NO intermediates are essential in both pharmacologic (anesthetic) and ischemic preconditioning. Although the study of the cardioprotective effects of NO has centered on the sGC signaling pathway, emerging evidence now shows that sGC-independent effects also are important. The mitochondrion is a cellular hub for both preconditioning and NO signaling and therefore represents a point of convergence in cardioprotection. Currently, the mitochondria have emerged as targets for cardioprotection, i.e., protecting the heart against the detrimental effects of acute IR injury. It is well established that mitochondrial dysfunction lies at the heart of cardiomyocyte death induced by IR injury. However, recent evidence indicates that the contribution of mitochondria to cell injury goes well beyond their role as death executors through the opening of mitochondrial PTPs, as they contain several specific and highly regulated intrinsic signaling pathways and molecular complexes that may be determinants for cell death or survival under stressful conditions. The field of cardiac mitochondrial biology continues to impress with various emerging novel concepts that will be highlighted in the future. We await with great anticipation the exciting developments in this field of research, which should result in the identification of novel mitochondria-targeted therapeutic strategies for treating cardiac disease.

 XML Download
XML Download