

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Tuberculosis still remains an important public health problem in Korea, where the prevalence of active pulmonary tuberculosis per 100,000 has decreased from 5,065 to 1,032 in the last 30 years.1 In addition, non-tuberculous mycobacteria (NTM) are increasingly encountered in the clinical laboratory, due to the dissemination of HIV, the increased use of immunosuppressive drugs, and the improvement in detection techniques such as liquid cultures.2 The correct and rapid diagnosis of mycobacteria to the species level is extremely important because the treatment of tuberculosis differs from the treatment of diseases caused by NTM, and adequate therapy taken in time can reduce the spread of the disease and the costs of hospitalization.
Since it was first introduced, the BACTEC 460 TB radiometric system (Becton Dickinson, USA) has been the gold standard for rapid detection of mycobacteria by use of the BACTEC 12B liquid medium containing 14C-labeled palmitic acid substrate. On the basis of CO2 evolution, this system has increased recovery rates and has decreased the time to detection of mycobacteria from 8 weeks with conventional cultures to 2 weeks. The BACTEC MGIT 960 system (BD, USA), the newer broth-based method that has recently been developed, offers rapid time to detection similar to that of the BACTEC 460 TB system while using fully automated, high-capacity, noninvasive, nonradiometric, and self-contained incubator readers.2 However, these broth-based systems lack appropriate diagnostic tests for rapid differentiation of different species of mycobacteria. Additional techniques such as high-performance liquid chromatography,3 Gen-Probes,4-6 and sequencing based on the 16S rRNA gene7,8 can provide rapid differentiation of a wide variety of mycobacteria growing in liquid cultures. However, these procedures require additional expensive equipment and remain limited to reference laboratories or they require several probes and cover a limited number of mycobacterial species.
Recently, polymerase chain reaction (PCR) techniques have provided new possibilities for the rapid identification of mycobacteria growing in liquid cultures. A wide variety of species-specific sequences, which can be candidate targets for single PCR to differentiate mycobacteria, have been reported in mycobacteria as follows: MPB70 in M. tuberculosis complex,9,10 DT6 in M. avium,6,11 DT1 in M. intracellulare,6,11 p6123 in M. kansasii,12,13 superoxide dismutase gene in M. fortuitum,14 16S-23S rRNA internal transcribed spacer region in M. gordonae,15 IS1395 in M. xenopi,16 and 16S rRNA hypervariable regions in M. terrae and M. chelonae.7 Considering the relatively large number of mycobacterial species-specific target genes presently characterized, at least 8 or more separate PCR reactions are required for identification of mycobacterial species isolated from liquid cultures by using a conventional PCR procedure. This is potentially expensive and labor-intensive and increases the risk of cross-contamination.
To overcome these limitations and to develop a rapid and reliable method to identify most common clinical mycobacterial isolates growing in liquid cultures, 2-step multiplex PCR assays were designed. In the first step, a multiplex PCR-A was initially used for detection of all mycobacterial species and differentiation of M. tuberculosis complex from NTM species. In the second step, 2 parallel multiplex PCR-B and PCR-C assays were used to allow simultaneous detection and identification of 7 different mycobacterial species such as M. avium, M. intracellulare, M. kansasii, M. fortuitum, M. gordonae, M. chelonae/M. abscessus, and M. terrae.
Materials and Methods
1. Reference strains and clinical isolates
Clinical mycobacterial isolates (n=132, Table 1) were retrieved from laboratory collections obtained between 1994 and 2005 at the Department of Laboratory Medicine, Chonnam National University Hospital (Gwangju, Korea). Fifteen mycobacterial reference strains, listed in Table 1, were included in this study. The reference strains were obtained from the Korean Institute of Tuberculosis (KIT), the Korean National Tuberculosis Association (KNTA), Seoul, Korea. Reference strains and clinical isolates were subcultured in MGIT tubes (BD), and the MGIT tubes were read by using the BACTEC MGIT 960 system (BD). When the tubes give positive signals, an aliquot was withdrawn from the tubes for acid-fast bacilli (AFB) staining. The AFB-positive isolates were subjected to DNA extraction.
2. Multiplex PCR assay
Mycobacterial DNA was extracted from MGIT culture media by using the heat lysis method as described previously.17 The multiplex primers were designed according to the recommendations described previously.18 Table 2 lists the combinations of primer sets for the multiplex PCR assays. For all multiplex PCR and species-specific PCR, amplification was performed in a DNA thermal cycler (GeneAmp PCR 9600; Perkin-Elmer, USA). A 5 µL aliquot of each DNA sample was used in a total volume of 50 µL PCR mixture containing 10 mM Tris-HCl (pH 8.8), 50 mM KCl, 1.5 mM MgCl2, 0.1% Triton X-100, 0.2 mM dNTPs, optimal amounts of primers (12.5~25.0 pmol), and 1 U of DynaZyme DNA polymerase (Finzymes, Oy, Finland). The multiplex PCR conditions were optimized as follows: in the first cycle, denaturation at 95℃ for 5 min; 35 cycles consisting of denaturation at 94℃ for 15 sec, annealing at 65℃ for 15 sec, and extension at 72℃ for 30 sec; and in the final cycle, extension at 72℃ for 10 min. Electrophoretic separation of each PCR product (10 µL) was performed in 2% agarose gel slabs in Tris-borate buffer [0.5×TBE; 0.04 M Tris-borate (pH 8.4), 1 mM EDTA] at 100 V for 30 min. The gels were stained in ethidium bromide solution (0.5 µg/mL) and the products were visualized in a transilluminator under UV light. Reference strains were run as positive controls, and deionized water was used as a negative control. For comparison of product lengths, 1 µg of 100 bp ladder (Bio-Rad, USA) was used as a molecular size marker.
3. PCR restriction fragment length polymorphism analysis (PRA) of hsp65
PCR-restriction enzyme analysis of the hsp65 gene (PRA-hsp65) was performed by the following modification of the original procedure as described previously.19 Briefly, an aliquot (5 µL) of each template DNA was used in 50 µL of the PCR mixture containing 10 mM Tris-HCl (pH 8.8), 50 mM KCl, 1.5 mM MgCl2, 0.1% Triton X-100, 0.2 mM dNTPs, 12.5 pmol of each primer TB11 (5'-ACCAACGATGGTGTGTCCAT-3') and TB12 (5'-CTTGTCGAACCGCATACCCT-3'), and 2 U of DynaZyme DNA polymerase (Finzymes). The amplification was performed as follows: in the first cycle, denaturation at 95℃ for 5 min; 35 cycles consisting of denaturation at 94℃ for 30 sec, annealing at 60℃ for 30 sec, and extension at 72℃ for 60 sec; and in the final cycle, extension at 72℃ for 10 min. For BstEII (Promega, USA) and HaeIII (Promega) digestions, 10 µL of each PCR product was added to a mixture containing 1 µL (20 U) of each enzyme, 3 µL of restriction buffer, and 16 µL of deionized water. The mixture was incubated for 60 min at 60℃ for BstEII digestion and 37℃ for HaeIII digestion. Electrophoretic separation of digested products was performed for 60 min at 100 V on 3% agarose gels, which were photographed under UV light after staining with ethidium bromide. Isolates were identified by using the PRA algorithm as described previously.20
Results
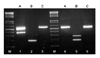
In order to validate the performance of the multiplex PCR assays for the detection and identification of members of the genus Mycobacterium and the differentiation of mycobacteria to the species level, a 2-step approach was performed for the reference strains listed in Table 1. In the first step, to distinguish M. tuberculosis complex from NTM, sample DNA was amplified by multiplex PCR-A with the primer pairs (A) listed in Table 2. In the second step, to differentiate NTM to the species level, DNA extracts were amplified by multiplex PCR-B and PCR-C in 2 parallel reactions by using each set of primer pairs (B and C) listed in Table 2. As shown in Fig. 1, multiplex PCR-A amplified 2 products of ~590 bp and 385 bp from M. tuberculosis ATCC 27294, whereas it amplified only a single ~590 bp product from NTM species. The multiplex PCR-B produced 648 bp, 347 bp, 275 bp, and 187 bp bands from M. intracellulare, M. kansasii, M. fortuitum, and M. avium reference strains, respectively. The multiplex PCR-C was able to identify reference strains as M. chelonae/M. abscessus, M. gordonae, or M. terrae according to their product sizes of ~590 bp and 441 bp, ~590 bp and 221 bp, or ~590 bp and 149 bp, respectively (Fig. 1).
We tested the multiplex PCR assay on 147 liquid cultures of mycobacteria, which included 15 reference mycobacterial species. To determine and verify a specific gene amplicon by the multiplex PCR assays, each species-specific PCR and PRA-hsp65 were performed by using the same reaction conditions as for the multiplex PCR. The results obtained from the clinical isolates and reference strains by using the multiplex PCR, species-specific PCR, and PRA-hsp65 are shown in Table 3. All mycobacteria subjected to the multiplex PCR exhibited a positive Mycobacterium genus amplicon of ~590 bp. Of the 147 cultures, 143 yielded 1 isolate, whereas 4 grew 2 mycobacterial species. In 129 of the 143 culture tubes yielding a single isolate, the multiplex PCR results were equivalent as determined by single PCR analysis and PRA-hsp65 except for 8 M. intracellulare isolates. These isolates were not amplified by multiplex PCR nor by species-specific PCR with DT1 primers for M. intracellulare. For the other 6 cultures yielding no amplicon by the multiplex PCR, PRA-hsp65 identified the following species not represented by the multiplex PCR primer sets: M. marinum ATCC 927, 2 M. marinum clinical strains, M. scrofulaceum ATCC 19981, M. smegmatis ATCC 19420, and M. szulgai ATCC 35799. The multiplex PCR revealed 2 mycobacterial species in the remaining 4 mixed cultures (Table 3). The mixed cultures from patients who reported a history of repetitively treated pulmonary tuberculosis grew M. tuberculosis complex plus M. avium (n=3) or M. avium plus M. fortuitum (n=1) (Fig. 2). However, these cultures were unidentified or misidentified as a single species by PRA-hsp65.
Discussion
This Study Shows That The Advantages Of Multiplex Pcr Are Considerable, As Compared With Other Recent And Innovative Molecular Procedures, Such As Techniques For Analyzing Pcr Products By Sequencing Analysis,8,21 Restriction Enzyme Analysis,19,20,22,23 Or Hybridization Assays.8,24,25 Among These Methods, PRA-hsp65 As Proposed By Telenti Et Al.19 Is The Best Approach For Differentiating A Wide Variety Of Mycobacterial Species, But It Is Time-consuming And It Is Difficult To Read The Results Visually. In Addition, PRA-hsp65 Is Not Very Sensitive And It Cannot Detect The Coexistence Of Different Mycobacterial Species In Liquid Cultures. Among 132 Clinical Isolates In This Study, 6 Were Not Identified By PRA-hsp65 Because Of Mixed Cultures Of 2 Different Mycobacterial Species And Novel Pra Patterns Not Present In The Reference Algorithm.
In contrast, the multiplex PCR assays of this study and the study of Klemen et al.15 could differentiate these different isolates even when 2 or 3 mycobacterial species were present in a sample. Moreover, the method can be extended easily. As soon as specific DNA sequences are identified in other species of mycobacteria, a protocol can be formulated and another primer set for a multiplex PCR can be added. The insertion sequences IS1395 and IS2404/2406 were found in M. xenopi and M. ulcerans, respectively.16,26,27 These may be candidates for targets in a multiplex PCR because the entire procedure, including sample preparation, can be completed within one working day. It is also more rapid and easy to interpret the results than PRA-hsp65.
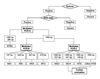
However, there are 2 limitations of the multiplex PCR-B. First, there is no primer for the 16S rRNA gene as a common mycobacterial positive control primer in the primer mixture of multiplex PCR-B, whereas such a primer pair for the 16S rRNA gene is included in the primer mixture of multiplex PCR-A and PCR-C. To overcome this limitation, the multiplex PCR-B should be performed in parallel with multiplex PCR-C. Second, the multiplex PCR-B assay has a low sensitivity for detection of M. intracellulare. The data for M. intracellulare showed that the multiplex PCR-B detected only 21 of 29 cultures (72.4%), indicating that the failure was due to the absence of the target gene DT1 in some clinical isolates of M. intracellulare or PCR inefficiency caused by a relatively large amplicon size. We performed species-specific PCR using DT1 primers for those 8 M. intracellulare isolates. The species-specific PCR also failed, revealing that there was no DT1 gene in those isolates. A study in Guinea-Bissau, West Africa, reported that none of 5 isolates identified as M. intracellulare by 16S rRNA sequencing was DT1-positive.28 Our study as well as that of Koivular et al.28 contradict the earlier reports6,11 on the usefulness of DT1 PCR as an identification method for M. intracellulare. Although the multiplex PCR-B using DT1 primers was unable to detect some M. intracellulare isolates in cultures, this problem was solved by another approach such as PRA-hsp65 because these isolates were checked by the multiplex PCR-C and underwent further evaluation according to the algorithm shown in Fig. 3.
To identify mycobacterial species growing from liquid media by use of these multiplex PCR assays, we suggest the detection algorithm shown in Fig. 3. The first step is to culture the mycobacteria with the BACTEC MGIT 960 system. If the cultures are positive in the system, proceed to the next step. The next step is to perform AFB staining. If AFB is present, proceed to multiplex PCR-A. If AFB are absent and the samples are contaminated with other bacteria, discard the samples. The third step is to perform the multiplex PCR-A for differentiation of M. tuberculosis complex from NTM. If both 385 bp and ~590 bp (common) bands or only 385 bp bands are present, the results are M. tuberculosis complex. If only the common band is present, proceed to the multiplex PCR-B and PCR-C. The forth step is to perform the multiplex PCR-B and PCR-C. If each specific band is present or if both specific and common bands are present, the results are M. avium, M. intracellulare, M. kansasii, M. fortuitum, M. gordonae, or M. terrae except M. chelonae. If only the common band is present, the result is other NTM and proceed to further evaluation. If the M. chelonae band is present, proceed to further evaluation. The multiplex PCR-C is unable to discern between these 2 species, because M. chelonae shares the primer sequence of 16S rRNA hypervariable region with M. abscessus for the multiplex PCR-C assay (Fig. 1).
In conclusion, the multiplex PCR assays in the present study may be an easy and reliable method for the rapid detection and differentiation of commonly encountered mycobacterial species in liquid cultures, even in those in which different species coexist.




 XML Download
XML Download