

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Diagnosis of hypertrophic cardiomyopathy (HCM) and left ventricular noncompaction cardiomyopathy (LVNC) is mainly based upon the morphological features. Failure to reach a clear diagnosis in suspected cases is not infrequent and is attributed to inapparent morphologic features. Assessing genetic abnormalities in such patients may be of limited help as our knowledge of the penetrance of genetic anomalies for cardiomyopathy is limited.
Traditionally, HCM, restrictive cardiomyopathy (RCM), dilated cardiomyopathy (DCM), and LVNC have been regarded as separate and distinct entities. However, recently a number of studies have demonstrated that the same genetic abnormality may result in diverse phenotypes. Therefore, HCM, DCM, RCM, or LVNC may be manifested by a single gene mutation. However, mixed features in a single patient have not been well appreciated.
Case
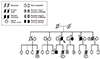
Molecular genetic testing was performed using a next-generation sequencing (NGS)-based targeted gene panel consisting of 25 known hypertrophic cardiomyopathy-related genes. A novel missense mutation in the troponin I3 (TNNI3), Arg186Gly, was identified, which was confirmed by Sanger sequencing. The family pedigree is shown in Fig. 1.
Case 1
Proband visited our hospital because of exertional dyspnea, and had a past medical history of cerebral infarction. Echocardiographic features were suggestive of RCM. As the prior echocardiogram was not available for comparative purpose, presence of previous different morphological features could not be ascertained. After cardiac transplantation in this patient, histological examination of the explanted heart revealed interstitial fibrosis, suggesting the possibility of idiopathic RCM. The patient was admitted again nine years after the cardiac transplantation for fulminant hepatitis, which ultimately led to death.
Case 2
Initial echocardiogram performed 13 years ago, showed only mild anterolateral wall hypertrophy (end-diastolic thickness of 15 mm) without prominent trabeculations at the apex. An echocardiographic examination performed nine years later showed septal hypertrophy as well as increased anterolateral wall hypertrophy (end-diastolic thickness of 20 mm). The echocardiogram also demonstrated prominent trabeculations at the apex suggestive of inapparent form of LVNC (Fig. 2). An initial echocardiography showed a marked relaxation abnormality that persisted until the most recent follow-up. Among her children, including a son and a daughter, only the son shared the same genetic mutation. However, the echocardiogram was not suggestive of any cardiomyopathy in this case.
Case 3
The patient's chief complaint was dyspnea, and he was referred to our hospital with a diagnosis of HCM by another hospital. Endomyocardial biopsy performed at the previous hospital revealed histological findings suggestive of HCM. Echocardiography performed at our hospital revealed asymmetric septal hypertrophy that was not as prominent as usually seen in typical patients with HCM (Fig. 3). Four years later, he was admitted to the neurology department with a diagnosis of basal ganglia infarction. The cardiac rhythm was predominantly in atrial fibrillation, with the occasional conversion to sinus rhythm lasting less than a day. The echocardiogram showed hemodynamic features of RCM that persisted even when the heart had sinus rhythm (Fig. 4).
Case 4
This patient was screened for the presence of HCM at the age of 18 after his younger brother's sudden death. The echocardiogram was normal with no features suggestive of HCM. When echocardiogram was repeated at the age of 35, it showed mild localized area of basal anterolateral wall hypertrophy (15 mm in thickness) and inappropriate diastolic function for his age. Moreover, a few prominent trabeculations were observed at the apex (Fig. 5). In this patient, implantable cardioverter defibrillator was considered but was declined by the patient as he had observed all the sudden deaths in his family occurred before the age of 30.
Discussion
Since the first report of β-myosin heavy chain gene mutation in 1990,1) gene mutations coding nearly every component of sarcomere have been reported.2)3)4)5)6)7)8)9) Moreover, our recent understanding became more complex than the initial perspectives that one single gene mutation can manifest as diverse cardiomyopathies.10)
Cardiac troponin I (cTnI) is the inhibitory subunit of troponin complex, which is responsible for inhibition of actomyosin ATPase activity. Therefore, cTnI is a key regulatory protein in cardiac muscle contraction and relaxation cycle. There are 3 genes encoding 3 TnI protein isoforms. The slow skeletal TnI is the predominant isoform expressed within the fetal heart. This isoform declines rapidly after birth and is replaced by the cardiac isoform 3, which is encoded by the TNNI3 genes.
After the first report about cTnl mutation in HCM,6) various mutations in cTnl causing HCM, DCM and RCM were reported.11) In cTnI mutations causing HCM, genotype-phenotype relationship is quite diverse. In one study, Lys183 del mutation in the cTnI gene was found to be associated with high penetrance, sudden death at any age, and dilated cardiomyopathy like features in subjects aged over 40 years.12) In another study by Mogensen J et al.13) involving largest numbers of patients with cTnI mutation, clinical expressions of cTnI3 mutations was quite heterogeneous, varying within and between the families. As mutation was present in a thin filament, before accumulation of our experience with large cases of cTnI mutations, we expected cTnI mutation to show mild hypertrophy as has been reported for TnT mutations.4) However, in 23 families with 13 different mutations in the reported study, degree of hypertrophy was quite diverse. Among the reported mutations, a patient was diagnosed with Arg186Gln mutation at the age of 29, which was present at the same location but in different single base pair as compared to our patients. The patient showed severe asymmetric septal hypertrophy of 35 mm and developed LV wall thinning and dilation over 13 years, the clinical feature of which was quite different from our affected family members with Arg186Gly mutation. Hypertrophies in patients in our study were mild and not extensive. In addition, in two patients in our family, trabeculations with deep recesses were noted at a limited area of the apex. Although we cannot state that this finding is an unequivocal LVNC, we speculate whether these findings are a mild form of LVNC.
In 2003, Mogensen et al.14) reported cTnI mutation as etiological causes of RCM. In their report, some family members also harbored features suggesting HCM as well as RCM. Therefore, presentation of RCM in one patient and rapid transition from HCM to RCM seen in another patient in our study may essentially represent same features observed in the reported study. Interesting finding reported in their study was the post-mortem microscopic findings seen in one patient. The authors noticed myocyte hypertrophy and myofibrillar disarray similar to the findings in HCM patients. I n our patient, who showed rapid transition from HCM to RCM, endomyocardial biopsy was performed elsewhere before he presented to our hospital. Based on the biopsy report, interstitial fibrosis was not marked, whereas myofiber hypertrophy was noted. Therefore, they concluded that histological findings were in favor of HCM.
In the previous reports,12)13) sudden death occur in all age groups. However, in our family, all the sudden deaths occurred under the age of 30. Nobody with sudden cardiac death ever had echocardiography before cardiac death nor had postmortem examination. Therefore, we are not aware of the states of disease manifestation or the degree of hypertrophy in these family members with sudden cardiac death.




 XML Download
XML Download