

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Hypertension is the leading risk factor in cardiovascular disease and renal disease. A critical goal to manage hypertension in patients is to lower elevated blood pressure. Essential hypertension is recognized as an individual having high blood pressure with no identifiable cause and usually occurs in 95% of patients. Hypertension is a complex phenotype and is affected by advanced age, increased body weight, high dietary sodium intake, and genetic factors.
The spontaneously hypertensive rats (SHR) experimental animal model is the most representative of essential hypertension. SHR have systolic blood pressures that are higher than 200 mmHg. Chronic inhibition of nitric oxide synthase with l-arginine derivatives, such as L-NG-nitro-L-arginine methyl ester (L-NAME), induces hypertension.1) Establishing an L-NAME-induced hypertension model is a relatively easy technique. The renin-angiotensin-aldosterone system (RAAS) regulates blood pressure. Angiotensin II is a peptide hormone that causes vasoconstriction and leads to organ damage, including heart and kidney damage.2)
Histone deacetylases (HDACs) are enzymes that remove acetyl groups from histone or non-histone proteins. HDACs are divided into four families. Class I HDACs include HDAC1, 2, 3, and 8. Class IIa HDACs include HDAC4, 5, 7, and 9. Class IIb HDACs include HDAC6 and HDAC10. HDAC11 belongs to class IV HDAC. Expression of specific HDAC isoforms is closely associated with cancers. For example, expression of HDAC1 and HDAC2 is upregulated in hepatocellular carcinoma.3) HDAC3 protein levels increase in esophageal cancer cells and human colon tumors.4)5) HDAC7 expression levels are low in cancer cells.6)
It was recently reported that HDAC3 catalyzes acetylation of the mineralocorticoid receptor (MR) in order to regulate MR target gene expression.7) HDAC4 is involved in the stimulation of MR-dependent transcription8) and is implicated in hypertension through vascular inflammation.9) Furthermore, HDAC4 protein expression is more likely in brains from SHR than those from Wistar-Kyoto (WKY) rats.10)
The protein levels of HDAC1, HDAC2, and HDAC3 increased in a pulmonary hypertensive rat model induced by hypoxia.11) Angiotensin II induced phosphorylation of HDAC5, but not HDAC5 protein expression, in vascular smooth muscle cells.12) We, and others, have previously reported that HDAC6 protein levels and enzymatic activity are upregulated in deoxycorticosterone acetate (DOCA)-salt hypertensive rats.13)14) However, the protein expression of class I and class II a/b HDACs remains unknown in animal models of experimental hypertension.
Here, we investigated, for the first time, the expression patterns of class I and class II a/b HDACs in three animal models of hypertension. We suggest that dysregulation of HDAC expression may be associated with the development of hypertension.
Materials and Methods
Materials
Angiotensin II (CAS 4474-91-3) was purchased from Calbiochem (Merck Millipore Corporation, Darmstadt, Germany). L-NAME (sc-200333A) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
Spontaneously hypertensive rats
WKY (n=14) rats and SHR (n=14) were purchased from SLC (Shizuoka, Japan). Rats were housed in a temperature-controlled room with a 12 h light-dark cycle and free access to food and drinking water. The WKY rats and SHR were sacrificed at 8 weeks (n=6) and 24 weeks (n=8), respectively.
Angiotensin II-induced hypertensive mouse model
Male CD-1 mice (6 weeks old) were purchased from the Samtako Company (Seongnam, Korea). All animal experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (US National Institutes of Health Publication, 8th edition, 2011). Mice were housed in a temperature-controlled room with a 12 h light-dark cycle and free access to food and water. Mice were anaesthetized by intraperitoneal injection of ketamine (70 mg/kg) and xylazine (14 mg/kg). Angiotensin II (Ang II, 1.3 mg·kg-1·day-1) was infused into CD-1 mice (n=9) via an Alzet osmotic pump for 2 weeks.
L-NAME-induced hypertensive mouse model
To induce hypertension, male CD-1 mice (6 weeks old) were treated with L-NAME (0.1% in drinking water, n=7) for 2 weeks. Control mice were treated with tap water instead of L-NAME.
Blood pressures measurements
Blood pressure measurements were obtained as described previously.15) Briefly, the systolic blood pressure, diastolic blood pressure, and mean blood pressure were measured in conscious state by the tail-cuff method (Visitech Systems, BP-2000, Apex, NC, USA). For exact blood pressure measurements, the rats and mice were trained to adapt to the holder of the machine for 3 days before the start of the experiment. The systolic blood pressure value expressed is the average of 5 to 7 measurements.
Western blots
For western blot analysis, heart tissues were homogenized with a RIPA lysis buffer, as described previously.16) Proteins were subjected to 8% or 10% SDS-PAGE and transferred to a PVDF membrane. The membranes were incubated in a 5% skim milk blocking solution for 1 h and then with the following primary antibodies: anti-HDAC1 (06-720, Millipore), anti-HDAC2 (ab12169, Abcam), anti-HDAC3 (sc-11417, Santa Cruz), anti-HDAC4 (sc-11418, Santa Cruz), anti-HDAC5 (sc-133225, Santa Cruz), anti-HDAC6 (#7612, Cell Signaling), anti-HDAC7 (3607-100, BioVision), anti-HDAC8 (ab137474, Abcam), anti-HDAC9 (3609-100, BioVision), anti-HDAC10 (ab53096, Abcam), and anti-GAPDH (sc-32233, Santa Cruz). The blots were incubated with anti-rabbit or anti-mouse horseradish-peroxidase-conjugated secondary antibodies for 1 h. The blots were developed using Immobilon™ Western Detection Reagents (Millipore, Billerica, MA, USA).
Results
Elevated blood pressures in hypertensive animal models
We evaluated the blood pressure of three hypertensive animal models. SHR was the representative model for essential hypertension. At 24 weeks of age, SHR showed higher systolic (206.6±6.6 vs. 130.3±5.7 mmHg), diastolic (123.6±24.2 vs. 77.0±12.0 mmHg), and mean blood pressure values (151.6±18.0 vs. 94.9±8.2 mmHg) than the WKY rats (Fig. 1A-C). Ang II, a component of RAAS, was infused for 2 weeks to induce hypertension in the animals. The systolic blood pressure was higher in Ang II-induced mice (183.6±13.7 mmHg) than in sham mice (130.0±10.0 mmHg). The Ang II-induced mice also showed higher diastolic (139.2±12.5 vs. 92.0±11.4 mmHg) and mean blood pressure values (153.8±11.4 vs. 104.5±9.9 mmHg) (Fig. 1D-F).
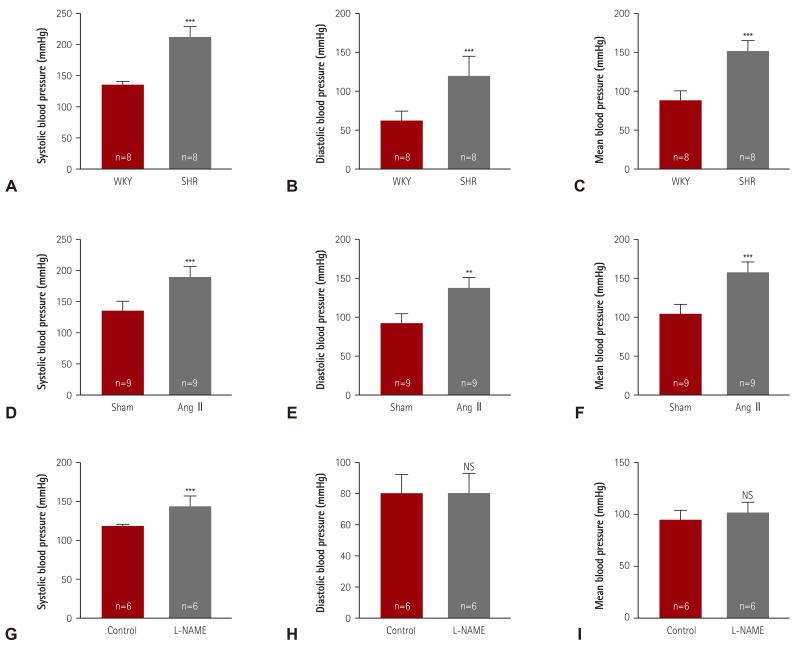
Fig. 1
Elevated blood pressures in hypertensive animal models. (A-C) Systolic, diastolic, and mean blood pressures were measured in a conscious state by the tail-cuff method. Twenty-four-week-old SHR and WKY rats were analyzed (n=8 per group). (D-F) After 2 weeks of angiotensin II (Ang II) infusion in mice, blood pressures were determined (n=9 per group). (G-I) Hypertension was induced by treatment with L-NG-nitro-L-arginine methyl ester (0.1% L-NAME, in drinking water) for 2 weeks, after which, the blood pressures were evaluated. **p<0.01 and ***p<0.001 vs. WKY, sham, or control group. SHR: spontaneously hypertensive rats, WKY: Wistar-Kyoto, NS: not significant, L-NAME: L-NG-nitro-L-arginine methyl ester.
![]()
L-NAME, a nitric oxide synthase inhibitor, induced a higher systolic blood pressure (150.3±8.0 vs. 123.7±2.6 mmHg) in treated mice than in sham mice, but did not affect the diastolic blood pressure or mean blood pressure (Fig. 1G-I).
Class I and II a/b HDAC expression in spontaneously hypertensive rats
Western blot analysis was performed to determine whether the HDAC expression profile in essential hypertension is changed between 8 and 24 weeks of age. In SHR, 8 weeks of age would definitely be an early time for demonstrating high blood pressure. At 24 weeks of age, considerably high blood pressure would be maintained. First, we examined the class I HDAC protein levels in the left ventricular tissue. At 24 weeks of age, the protein levels of HDAC1 and HDAC2 were not altered (Fig. 2A). However, HDAC3 expression was increased and HDAC8 expression was decreased in SHR. Unlike at 24 weeks of age, the HDAC1, HDAC2, and HDAC3 protein expression levels were significantly decreased in SHR at 8 weeks of age compared with the levels in WKY rats, whereas the HDAC8 protein levels were unchanged (Supplementary Fig. 1A, B in the online-only Data Supplement). Next, we examined class IIa HDAC protein expression. The expression of HDAC4, HDAC5, and HDAC7 was significantly higher in SHR than in WKY rats at 24 week of age. SHR did not show increased levels of HDAC9 protein (Fig. 2D). However, the HDAC5 and HDAC7 protein expression levels were significantly reduced at 8 weeks of age in SHR compared with the levels in WKY rats (Supplementary Fig. 1C, D in the online-only Data Supplement). The HDAC4 and HDAC9 protein expression levels were not changed at 8 weeks of age. In the case of the class IIb HDACs, the HDAC6 and HDAC10 protein levels were significantly higher in SHR than those in WKY rats at 24 weeks of age. However, HDAC6 protein expression was lower in SHR than in WKY rats at 8 weeks of age, whereas HDAC10 protein expression was not altered (Supplementary Fig. 1E, F in the online-only Data Supplement).
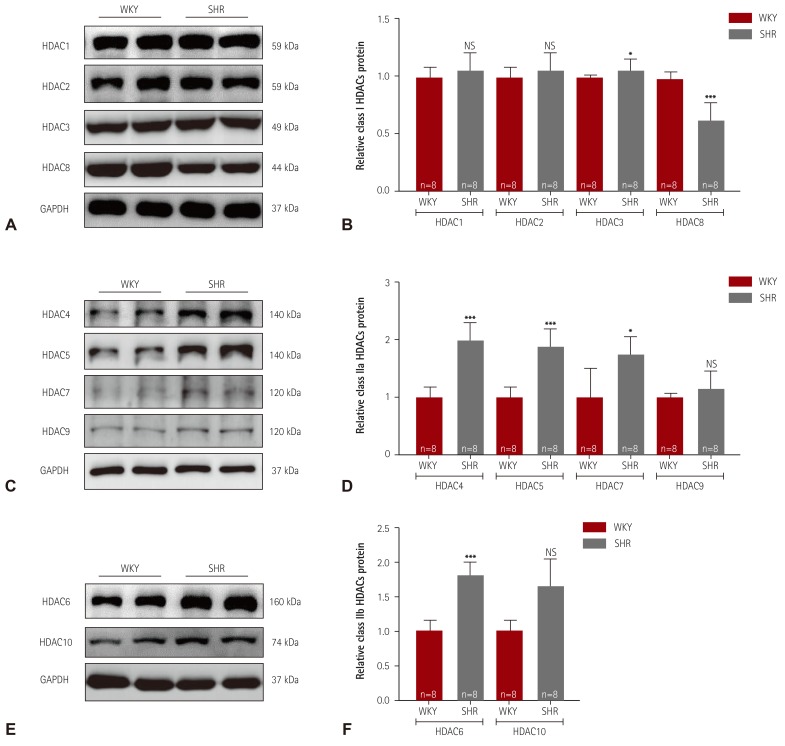
Fig. 2
Class I and IIa/b HDACs expression in spontaneously hypertensive rats. (A) Levels of class I HDACs (HDAC1, 2, 3, and 8) protein expression were determined by western blot analysis using left ventricular tissues from WKY rats and SHR. GAPDH was used as a loading control. (B) Proteins were quantified by densitometry. (C) Levels of cardiac class IIa HDACs (HDAC4, 5, 7, and 9) proteins were assessed in SHR by western blot analysis. (D) Protein levels were quantified by densitometry. (E) Levels of cardiac class IIb HDACs (HDAC6 and HDAC10) proteins were determined by western blot. (F) Quantification of class IIb HDACs. *p<0.05, **p<0.01, and ***p<0.001 vs. WKY. WKY: Wistar-Kyoto, SHR: spontaneously hypertensive rats, HDAC: histone deacetylase, NS: not significant, GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
![]()
Decreased expression of HDAC8 in angiotensin II-induced hypertensive mice
We examined class I HDACs protein expression in Ang II-induced hypertensive mice. As shown in Fig. 3A, B, HDAC8 protein expression was significantly lower in Ang II-induced mice than in sham mice. Expression of HDAC1, HDAC2, and HDAC3 was not altered in response to Ang II. We next examined class IIa HDACs. Expression of HDAC4, HDAC5, HDAC7, and HDAC9 was not different between the sham and Ang II-induced mice (Fig. 3C, D). We examined expression of class IIb HDACs in hearts from Ang II-induced mice. HDAC6 and HDAC10 protein expression was not altered in Ang II-induced mice (Fig. 3E, F).
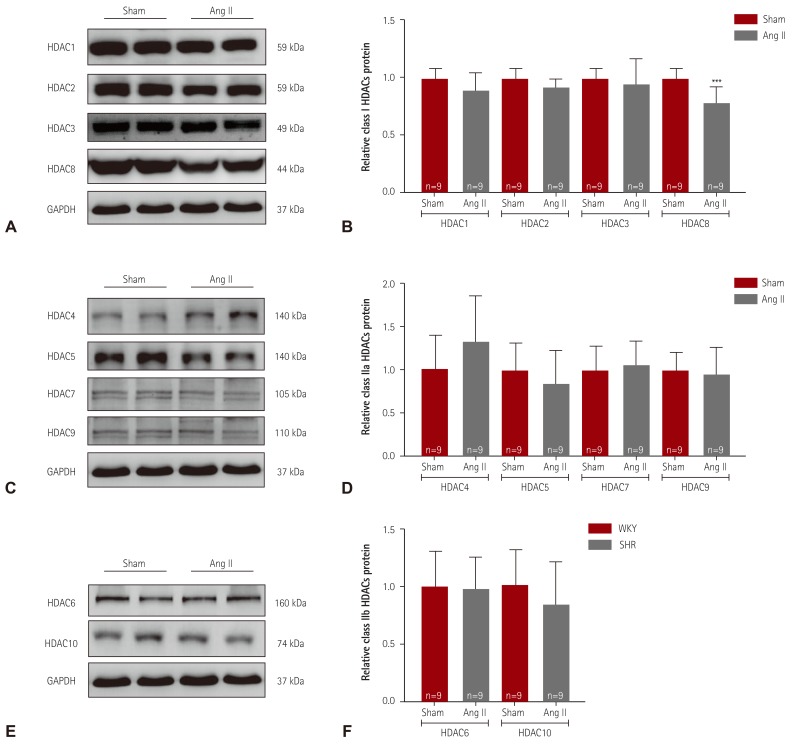
Fig. 3
Decreased expression of HDAC8 in angiotensin II-induced hypertensive mice. (A) Levels of class I HDACs (HDAC1, 2, 3, and 8) protein expression were determined by western blot analysis using hearts from angiotensin II (Ang II)-induced hypertensive mice and sham mice. GAPDH was used as a loading control. (B) Proteins were quantified by densitometry. (C) Levels of cardiac class IIa HDACs (HDAC4, 5, 7, and 9) proteins were assessed in Ang II-induced mice by western blot analysis. (D) Protein levels were quantified by densitometry. (E) Levels of cardiac class IIb HDACs (HDAC6 and HDAC10) proteins were determined by western blot. (F) Quantification of class IIb HDACs. ***p<0.001 vs. sham mice. HDAC: histone deacetylase, GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
![]()
Decreased class IIa HDAC expression in L-NAME-induced hypertensive mice
We investigated the HDAC expression profile in another experimental hypertension model, L-NAME-induced hypertensive mice. Hypertension was induced in mice by administration of L-NAME in the drinking water for 2 weeks. We performed western blot analysis to examine the expression of class I HDACs. As shown in Fig. 4A, B, we observed no differences between sham and L-NAME mice. Expression of all class IIa HDACs (HDAC4, 5, 7, and 9) was significantly lower in L-NAME mice than in sham mice (Fig. 4C, D). Furthermore, the protein levels of HDAC6 and HDAC10 in hearts from L-NAME mice were not different from those in sham mice (Fig. 4E, F).
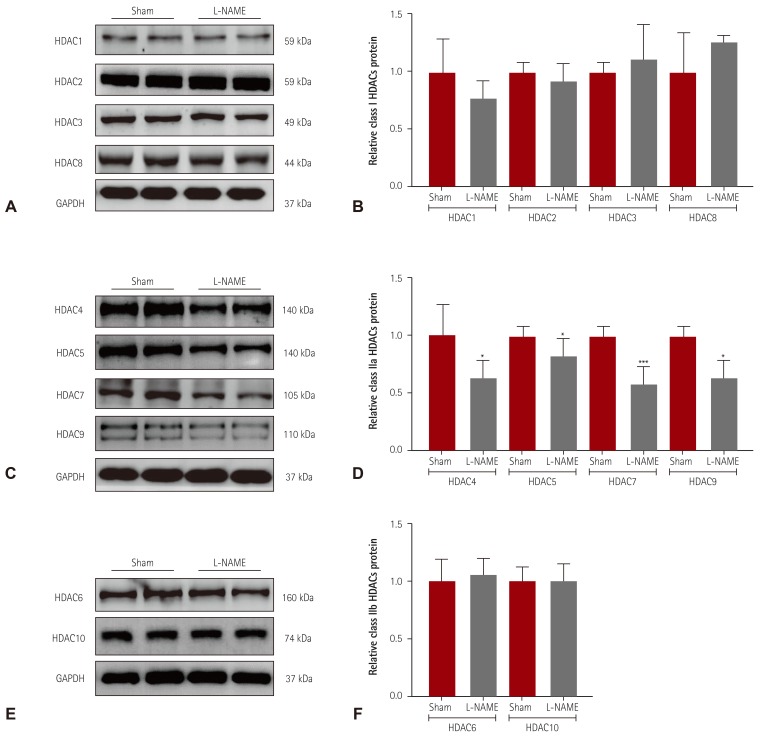
Fig. 4
Decreased class IIa HDAC expression in L-NAME-induced hypertensive mice. (A) Levels of class I HDACs (HDAC1, 2, 3, and 8) protein expression were determined by western blot analysis using hearts from L-NAME-induced hypertensive mice and sham mice. GAPDH was used as a loading control. (B) Proteins were quantified by densitometry. (C) Levels of cardiac class IIa HDACs (HDAC4, 5, 7, and 9) proteins were assessed in L-NAME mice by western blot analysis. (D) Protein levels were quantified by densitometry. (E) Levels of cardiac class IIb HDACs (HDAC6 and HDAC10) were determined by western blot. (F) Quantification of class IIb HDACs. *p<0.05 and ***p<0.001 vs. sham mice. L-NAME: L-NG-nitro-L-arginine methyl ester, GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
![]()
Discussion
The present study shows that dysregulation of HDAC is associated with systemic hypertensive animal models. SHR are the most commonly used experimental hypertension models that reflect human essential hypertension. Angiotensin II is a component of RAAS, which regulates blood pressure. We demonstrated, for the first time, that expression of class I or class IIa/b HDACs proteins changed in hypertensive mice and rats. The standards of the present study considered 6-month-old SHR as relatively old rats. Blood pressure measurements indicated that SHR developed high systolic blood pressure, with more than 200 mmHg.
In the present study, we found that the HDAC expression profile was switched to being age-dependent in SHR and WKY rats. From 8 to 24 weeks of age, the expression of most of the class I HDACs was repressed, whereas that of the class IIa/b HDACs had increased. In order to accurately identify the HDAC expression patterns, an earlier time frame (e.g., 4 or 6 weeks) before the appearance of high blood pressure (8 weeks) needs to be investigated in the near future.
We observed that class IIa HDACs (HDAC4, 5, and 7) and class IIb HDAC (HDAC6 and 10) protein levels were increased in SHR when compared with WKY rats at 24 weeks of age. Unexpectedly, class IIa/b HDAC protein expression was not changed in angiotensin II (Ang II)-induced mice. Post-translational modification of HDAC4 and HDAC5 has been reported to be linked to hypertension. For example, HDAC5 was phosphorylated by protein kinase D in Ang II-induced hypertension.17) Usui et al.18) have reported that HDAC4 controls neointimal hyperplasia and mediates hypertension via vascular inflammation in SHR.9) They addressed the increased expression of HDAC4 messenger ribonucleic acid and the induction of phosphorylated HDAC4 in response to stimulation by inflammatory cytokines such as TNF-α in vascular smooth muscle cells. They reported that MC1568 (a class IIa HDAC inhibitor) prevented the increase in activation of HDAC4 in neointimal lesions through p38 mitogen-activated protein kinase/heat shock protein 27 signals. In our most recently published paper, we demonstrated that HDAC4 and GATA-binding factor 6 (GATA6) expression levels were increased in the kidney and thoracic aorta in Ang II-induced hypertensive mice.19) We also showed that MC1568 treatment lowered increased blood pressure and weakened the association between HDAC4 and calcium/calmodulin-dependent kinase IIα, which is implicated in vascular hypertrophy and hyperplasia. So far, HDAC4 seems to be closely related to the development of hypertension. HDAC3 has been reported to be implicated in mineralocorticoid receptor-associated hypertension.7) HDAC6, a class IIb HDACs, has a structure similar to HDAC10. Two recent reports showed that HDAC6 protein expression and its catalytic activity were induced in chronic DOCA-salt hypertensive rats.13)14) In the present study, the increase in HDAC6 expression was only observed in SHR, but not in both Ang II-induced mice and L-NAME hypertension mice. Aside from its increase in Ang II-induced hypertensive mice (mentioned above), GATA6 protein expression was also induced in both DOCA-salt hypertensive rats and in SHR at 24 weeks of age (unpublished data). Taken together, the results indicate that the GATA6 transcription factor may be related to the modulation of HDAC4 and HDAC6 in hypertensive experimental models. We need to examine whether the relationship between GATA6 and HDAC4/6 is essential to the development of hypertension.
In SHR, HDAC8, a class I HDAC, is significantly reduced in SHR comparative to WKY rats. Furthermore, HDAC8 protein was also reduced in Ang II-induced mice. So far, there is no evidence that HDAC8 is involved in Ang II-induced hypertension.
In the present study, 2 weeks of L-NAME treatment in mice induced mild hypertension, while longer treatments (6-8 weeks) in rats produced systemic hypertension.20)21) We found that most class II a/b HDACs were downregulated in hearts from L-NAME-induced hypertensive mice compared to sham mice. However, as addressed above, protein expression of class IIa/b HDACs was upregulated in SHR, but the differences between SHR and L-NAME-induced hypertension cannot be explained in the present study.
To date, there were no studies that investigated HDAC expression in systemic arterial hypertension. In the present study, it has been clearly demonstrated that HDAC expression patterns are different among the three different types of animal models. These findings suggest that different mechanisms can exist for different experimental models of hypertension. In addition, the distinction of HDAC expression could be from the degree of hypertension or due to a difference of animal species used (rat vs. mouse). Therefore, we need to apply different therapeutic strategies for hypertension treatment based on the pathological state of the hypertension.
However, it was reported that particular isoforms of HDAC are upregulated or downregulated in different cancers. For instance, expression of class II a/b and class IV HDACs was reduced in glioblastomas.22)
In conclusion, the present study shows, for the first time, that class II a/b HDACs protein expressions are upregulated or downregulated in SHR and L-NAME-induced hypertension hearts, respectively. The biological function of class IIa/b HDACs in the development of hypertension remains to be elucidated.
 XML Download
XML Download