

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Fabry disease is an X-linked lysosomal storage disease caused by decreased activity of α-Galactosidase A (αGal-A). If the disease is left untreated, the natural long-term course is associated with severe multi-organ dysfunction of the renal, cardiac and/or cerebrovascular systems.1) Therefore, before irreversible organ damage occurs, early diagnosis of Fabry disease is important. However, currently more than 650 known mutations are individually responsible for complete or partial deficient activity of the enzyme αGal-A in patients with Fabry disease.2)3) This deficient can lead to the wide variety of progressive clinical manifestations in affected individuals. In addition, the clinical spectrum of Fabry disease ranges from the classic form, which appears in early childhood, to the late-onset form, which is diagnosed in adulthood. The diagnosis of Fabry disease is difficult and takes a long time. In a previous study, the mean duration from the onset of symptoms to the correct diagnosis was 13.7 and 16.3 years in males and females, respectively.4)
Here, we report the case of a 39-year-old man who showed atypical chest discomfort symptoms with a classic form of Fabry disease caused by a novel galactosidase alpha (GLA) mutation (p.Leu206 Pro).
Case
The patient was a 39-year-old man when he visited an outpatient cardiologist due to intermittent and atypical chest discomfort symptoms and an abnormal electrocardiograph (T inversion in I,II, aVL, aVF, V4-6; left ventricular hypertrophy with voltage criteria; sinus bradycardia) (Fig. 1A). He received a transthoracic echocardiograph examination, and his echocardiograph demonstrated that his left ventricular (LV) systolic function was functioning normally. However, his interventricular septum thickness and LV posterior wall thickness had increased to 18.1 mm and 13.0 mm, respectively (LV mass index 203.9 g/m2; relative wall thickness, 0.61) (Fig. 1B). He did not display any cardiac arrhythmias, such as ventricular tachycardia, but mild bradycardia was observed using a Holter monitor. To evaluate the additionally functional and morphological changes of the myocardium, cardiac magnetic resonance imaging was performed. There were no abnormal findings of the heart on morphologic, functional, perfusion or viability studies. His physical examination revealed proteinuria (P/C ratio 0.501) without hematuria and normal renal function. He also experienced intermittent pain in his extremities which was associated with atypical chest discomfort, a ringing in the ears in the past, angiokeratomas on the trunk, and cornea verticillata. These findings suggested a clinical diagnosis of a classic form Fabry disease. We then measured αGal-A activity in his plasma using a fluorometric enzyme assay. The patient's plasma αGal-A activity was markedly lower than the mean value of the controls (4.1 nmoL/hr/mg protein versus 60.7 nmoL/hr/mg protein); the cutoff value that indicates below-normal αGal-A activity in plasma is 35 nmoL/hr/mg protein. After genetic counseling and obtaining written informed consent, we obtained genomic deoxyribonucleic acid (DNA) from the peripheral blood using a standard technique. We assessed all of the GLA exons and the flanking intronic regions by direct sequencing of polymerase chain reaction products. We identified one hemizygous mutation in exon 4 of GLA, c.617T>C (p.Leu206 Pro). The mutation was absent from the single nucleotide polymorphism database and H, G, V, D. Since the patient's mother's DNA was not available, it could not be confirmed that the mutation was de novo.
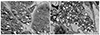
To evaluate the presence of organ impairment before enzyme replacement therapy, a renal biopsy and abdominal computed tomography (CT) was performed. Abdominal CT revealed a slightly small kidney with bilateral parenchymal thinning (right kidney 8.5×4.9 cm left kidney 7.6×5.9 cm). Renal histology of the left kidney showed tubular epithelial cells with a lacy, vacuolated cytoplasm. Interstitial macrophages and vascular endothelial cells also appeared to be vacuolated. Two glomerui were included in a frozen tissue sample for immunofluorescent staining and their podocytes had expanded cytoplasm that appeared pale and lacy. Mild lymphocytic infiltrates were present in the cortical interstitium without fibrosis. No deposits were identified in immunofluorescent staining. Ultra structure shown by electron microscopy revealed many electron-dense whorled, laminated inclusions filling the podocyte cytoplasm. These inclusions were composed of concentric dense layers (Fig. 2A, B). The inclusions were also present in mesangial, endothelial and tubular epitherlial cells in much smaller amounts. The patient was finally diagnosed as having Fabry disease with major organ dysfunction.
Discussion
Fabry disease is a devastating, progressive inborn error of metabolism associated with absent or deficient activity of αGal-A. In the early stages, the lack of enzyme αGal-A activity results in progressive accumulation of globotriaosylceramide (GL-3 or Gb3) and related glycosphingolipid deposition within lysosomes which are ubiquitous subcellular organelles in a variety of cell types.5) This leads to multi-organ cellular dysfunction and microvascular pathologic changes.6) However, during the very first years of life, most patients remain clinically asymptomatic. The first clinical symptoms interfering with a person's well-being and performance occur in childhood, typically between the age of 3 and 10 years, and generally a few years later in girls than in boys.7)8) With increasing age, progressive impairments to major organ systems develop in both genders, leading to organ failure. Life-threatening cardiovascular or cerebrovascular complications and end-stage renal disease limits the expectation of life of untreated males and females to approximately 50 and 70 years, representing reductions of 20 and 10 years, respectively, as compared to the general population.9)
In this case, the first clinical symptom to occur was in childhood consisting of a burning pain originating in the extremities and radiating inwards to the limbs. He and his family regarded the problem as ‘growing pains’. He also reported intermittent chronic pain characterized by burning and tingling paraesthesias. However, pain may wane in adults and it is important to search for medical history.10)
The estimated disease incidence ranges from 1 in 1500 to 1 in 476000 in the general population, according to newborn screening initiatives.11)12) Other reported incidences range from 1 in 1250 to 1 in 11700 live male births.13) The 14-kb αGal-A is located at Xq22.1 and consists of seven exons. The 1.45-kb GLA messenger ribonucleic acid encodes a polypeptide of 429 amino acids, including a 31 amino-acid N-terminal signal peptide. More than 650 mutations in GLA have previously been identified in Fabry disease patients. Recently, Umeda et al.3) reported a new GLA mutation (p.Phe69 Leu) in a Japanese patient with late-onset Fabry disease. In our case, we sought to determine whether the Leu206 Pro mutation in GLA-altered protein function contributed to Fabry disease using automated methods available on the internet, including the fabry-database (http://fabry-database.org/), Labgenomics GLA mutation database, the human gene mutation database, and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), but the genetic mutation could not be found on these sites.
According to the current proposed guidelines for stating enzyme replacement therapy (ERT) in Fabry disease patients,14) every male Fabry patient over 16 years old should be offered ERT, irrespective of the stage of chronic renal disease. There are currently two commercially available enzyme preparations for Fabry disease: agalsidase alfa (Replagal®; Shire, Cambridge, MA, USA), produced using cultured human skin fibroblasts and registered for use at a dose of 0.2 mg/kg biweekly, and agalsidase beta (Fabrazyme®; Genzyme Corp., Cambridge, MA, USA), produced by the expression of human a-galactosidase cDNA in Chinese hamster ovary cells and registered for a use of 1.0 mg/kg biweekly.15) It is hoped that long-term ERT can halt disease progression; however, the importance of adjunctive therapies should be noted.1)
In conclusion, a novel missense mutation in GLA was identified, Leu206 Pro, in a classic form Fabry disease patient. A 39-year-old male Fabry disease patient requires ERT and adjunctive treatment. It is hoped that long-term therapies can delay vital organ impairment. Furthermore, the identification of this novel mutation will enable clinicians and genetic counselors to have a better clinical understanding of Fabry disease.

 XML Download
XML Download