

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Cardiomyopathies (CMPs) are a heterogeneous group of heart muscle diseases associated with mechanical and/or electrical dysfunction, including hypertrophic cardiomyopathy (HCMP), dilated cardiomyopathy (DCMP), restrictive cardiomyopathy (RCMP), left ventricular noncompaction (LVNC) and arrhythmogenic right ventricular dysplasia.1) An increasing number of CMPs are now recognized to have familial forms, which result from single-gene mutations with a Mendelian inheritance pattern.2) However, the ability to identify disease-causing mutations is quite limited, because of the marked genetic and allelic heterogeneity and the lack of complete knowledge of the mutations that lead to CMPs. To date, more than 100 genes and more than 1400 genetic variations in sarcomere proteins have been identified as causative mutations of CMPs1)3) Another obstacle for the genetic diagnosis of CMPs is the realization that although they are clinically distinct entities, there is a genetic overlap among CMPS. For example, although mutations in cardiac troponin (cTn) are most commonly associated with HCMP and RCMP, they have also been reported in DCMP4) and LVNC5) Furthermore, a single mutation could cause not only a specific CMP, but also several different CMPs.6)
Troponin (Tn) is a critical regulator of muscle contraction in cardiac muscles, and is composed of three subunits: cardiac troponin I (cTnI), cardiac troponin C (cTnC), and cardiac troponin T (cTnT).4) cTnI can inhibit the actomyosin ATPase activity independently of the other Tn subunits. cTnI is encoded by the troponin I type 3 gene (TNNI3), which is a 5966-bp gene located on chromosome 19 consisting of eight exons and encoding a 210-amino acid protein.7)8) Allelic variants of TNNI3 have been implicated in HCMP, RCMP, and DCMP.4)
Recently, targeted gene panels based on a next-generation sequencing (NGS) platform has been used to analyze the exons and flanking intronic regions of genes associated with diseases of interest. Compared with whole-exome and whole-genome sequencing, targeted gene panels with a limited number of genes provide a higher depth of coverage with increasing sensitivity and specificity, and a higher capacity to interpret the findings in a clinical context.3) Here, we report a large family, in which the p.Arg145Trp mutation of TNNI3 causes diverse phenotypes ranging from RCMP to HCMP to near-normal heart, as demonstrated with the NGS-based gene panel testing.
Case
Clinical evaluation
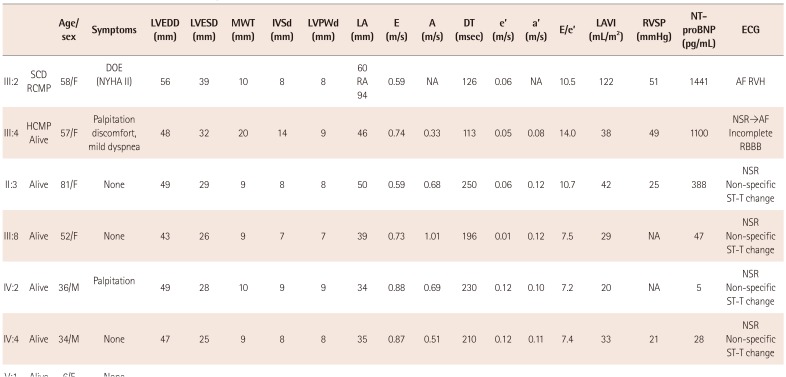
Clinical investigation of the proband and 13 family members was performed (Fig. 1, Table 1). A diagnosis of cardiomyopathy was made on the basis of clinical symptoms, physical as well as cardiologic examination including 12-lead electrocardiography (ECG), chest radiography, echocardiography, and whenever possible, chest contrast computed tomography (CT), cardiac magnetic resonance imaging (cardiac MRI), and laboratory studies including N-terminal of the prohormone brain natriuretic peptide (NT-proBNP). This study received the institutional review board approval (File No. 2015-07-029), and informed consent was obtained from all subjects.
NGS-based gene panel
A gene panel was designed to include 31 HCMP genes: ACTC1, ACTN2, ANKRD1, BAG3, CAV3, CRYAB, CSRP3, GLA, JPH2, LAMP2, LDB3, MYBPC3, MYH6, MYH7, MYL2, MYL3, MYLK2, MYOZ2, NEXN, OBSCN, PLN, PRKAG2, RYR2, TCAP, TNNC1, TNNC2, TNNI3, TNNT2, TPM1, TTR, and VCL. After obtaining informed consent, genomic DNA was extracted and captured with the Nextera Rapid Capture Custom Enrichment Kit (Illumina Inc., San Diego, CA, USA) and sequenced on a MiSeq platform. After screening all HCMP-related genes, we identified a missense mutation (c.433C>T, p.Arg145Trp) in the TNNI3 gene.
Case presentation
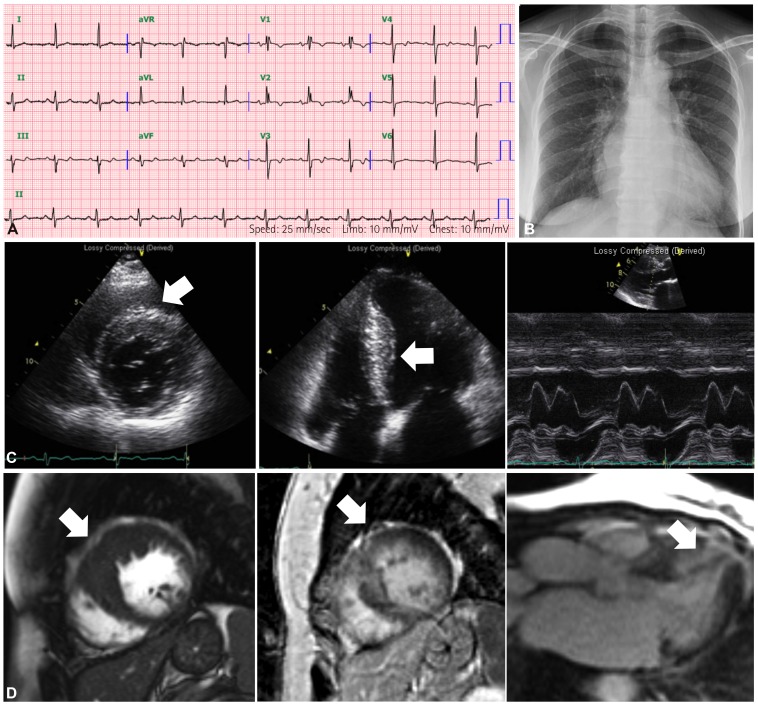
The proband (III:4) was a 57-year-old woman. At the age of 43 years, she was diagnosed for the first time with asymptomatic cardiomegaly (cardiothoracic ratio was 0.62) on chest radiograph (Fig. 2B), and later diagnosed with HCMP at another hospital. At the age of 51 years, she visited our hospital. Initially, ECG showed a normal sinus rhythm with incomplete right bundle branch block, left atrial enlargement and anterolateral T wave abnormalities (Fig. 2A). Echocardiographic examination revealed asymmetrical septal hypertrophy without left ventricle (LV) outflow tract obstruction, accompanied by left atrial enlargement, restrictive filling pattern, normal LV systolic function and normal cavity size (Fig. 2C). Cardiac MRI also showed delayed hyper-enhancement, which reflects fibrotic change in the myocardium in the hypertrophied segments (Fig. 2D). An arrhythmic event (atrial tachycardia), followed by an event of paroxysmal atrial fibrillation, was found on intermittent Holter monitoring. The patient's clinical features were compatible with those of HCMP.
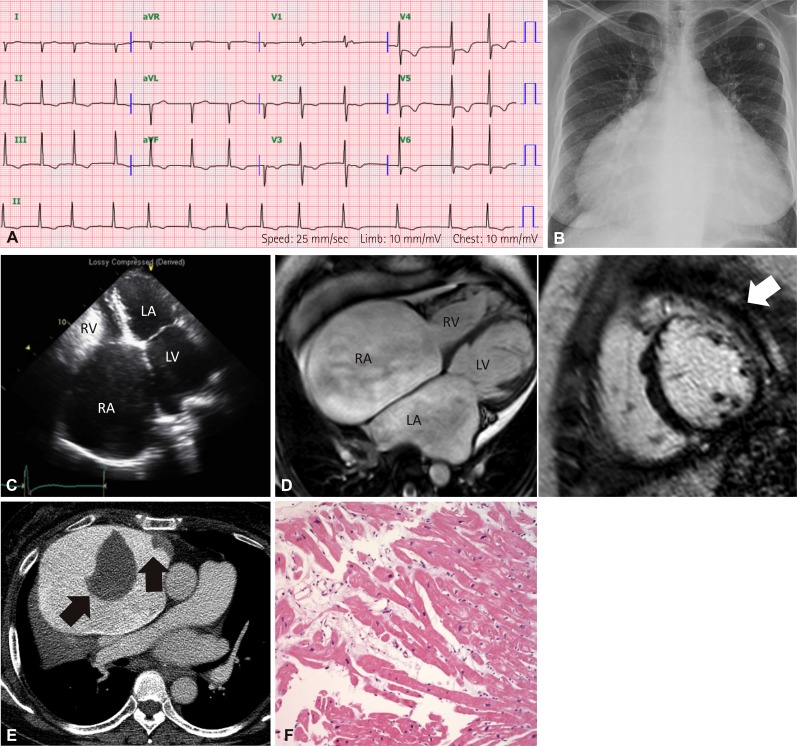
Family history taking revealed that the patient's older sister (III:2) also had cardiomyopathy. The sister had symptoms of heart failure from the age of 51. At the age of 54 years, she visited our hospital. ECG showed atrial fibrillation and ST-T abnormalities in inferolateral leads without low voltage in the limb leads (Fig. 3A). Severe cardiomegaly was noted in the chest radiograph (cardiothoracic ratio was 0.83) (Fig. 3B). Echocardiographic examination showed severe bi-atrial enlargement and a restrictive filling pattern accompanied by normal LV systolic function and wall thickness (Fig. 3C). In addition, cardiac MRI revealed mid and basal wall myocardial fibrosis from the basal to mid anterior, anterolateral and anteroseptal segments of the LV (Fig. 3D). Endomyocardial biopsy from the right ventricle showed nonspecific microscopic findings, such as mild focal disarray of myocytes with karyomegaly and interstitial widening (Fig. 3F). She succumbed to sudden death at the age of 58 while awaiting cardiac transplantation. Her last chest contrast CT, performed a few days before her death, revealed interval-increased extent of markedly dilated both atria, with two large thrombi in the right atrium (Fig. 3E). Her clinical features were typical of RCMP.
Family history revealed that a younger sister (III:5) of the proband had suddenly expired at the age of 21. Family genetic analysis further revealed five other affected members (II:3, III:8, IV:2, IV:4, V:1), and cardiac evaluation was performed for all family members, except one (V:1). Notably, the proband's mother (II:3) (81 years old) did show subtle cardiac abnormalities. Her ECG showed a mild degree of first degree AV block and non-specific ST-T changes in the inferolateral leads (Fig. 4A). Echocardiography showed a mild degree of left atrial enlargement with mild diastolic dysfunction (Fig. 4C). The other members (III:8, IV:2, IV:4) showed non-specific ST-T changes in ECG. Echocardiography revealed no significant abnormal findings except mild diastolic dysfunction in III:8.
Sanger sequencing of the TNNI3 gene mutation
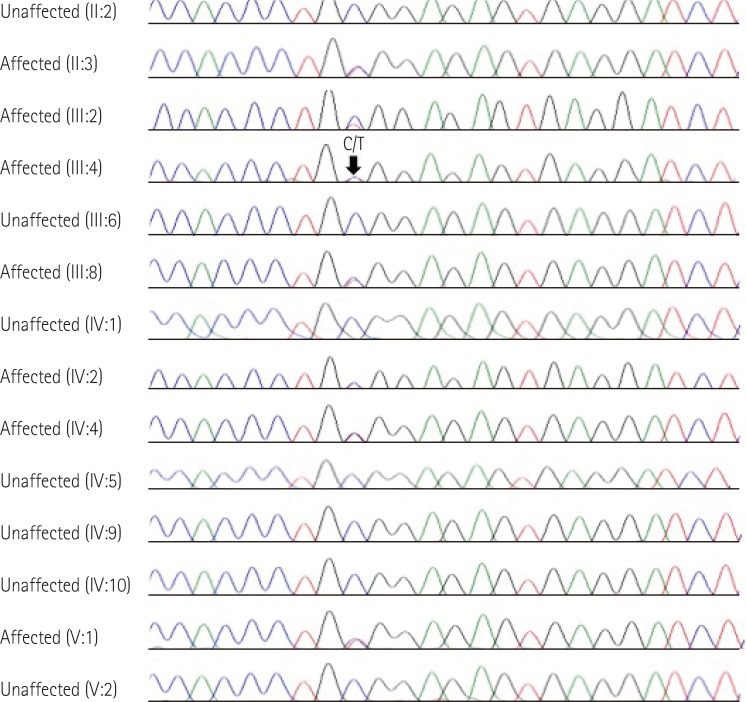
The TNNI3 mutation, p.Arg145Trp, was validated by Sanger sequencing in the proband (III:4) and her available family members (Fig. 5). To investigate the genetic diagnosis of the affected individual who had already died (III:2), genomic DNA was isolated from frozen heart tissue from an earlier biopsy. Exon 6 of TNNI3 was amplified using primer sets designed by the authors: TNNI3-e6-F, 5'-ggggattcagttccaggatt-3', TNNI3-e6-R, 5'-gcatttctgaggacccctta-3', TNNI3-e6-tissue-F, 5'-aggatggaggagttgggtgt-3', TNNI3-e6-tissue-R, 5'-aggtccagggactccttagc-3'. The PCR products were sequenced on the ABI Prism 3730xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) using the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems). DNA sequences were analyzed by comparison with reference sequence of TNNI3, NM_000363.4.
Results showed that the proband (III:4) was heterozygous for the missense mutation (c.433C>T; p.Arg145Trp) in the TNNI3 gene. The family study showed that her mother (II:3) carried the mutation, indicating maternal transmission. Her two sisters (III:2 and III:8), her second nephew (IV:2) and his daughter (V:2), and the proband's son (IV:4) also had the same TNNI3 mutation. According to a literature review and mutation database, the TNNI3 c.433C>T (p.Arg145Trp) mutation has been previously described in patients with RCMP,6) and its pathogenicity has been validated in vivo and in vitro by independent functional assays.9)10)11)
Discussion
In this report, we present a family carrying a TNNI3 mutation producing variable phenotypic expression. Although the TNNI3 c.433C>T (p.Arg145Trp) mutation has been reported in patients with isolated RCMP,4)6) no variable expressivity in a single family has been noted in such cases to date. It was previously thought that HCMP, DCMP, and RCMP were discrete and separate CMPs, but the finding of a single mutation in a sarcomere protein that is associated with phenotypic variability in CMPs, indicates that CMP comprises a spectrum of hereditary cardiac contractile protein diseases.4)6)
The cTnI is the inhibitory subunit of Tn, the thin filament regulatory complex that confers calcium-sensitivity to striated muscle actomyosin ATPase activity. Wild-type human cTnI inhibited ATPase activity in a Ca2+ concentration-dependent manner.9) It has been reported that allelic variants of TNNI3 mutation provoke HCMP (74%), RCMP (23%), and rarely DCMP (3%).4) The p.Arg145Trp mutation identified in our study occurs within the highly conserved inhibitory region, which is critical to the biological activity of TnI.12)13) Using actomyosin ATPase assays, Gomes et al.9) demonstrated that p.Arg145Trp has a reduced ability to inhibit ATPase activity in the absence of Ca2+ and, furthermore, that the mutant was unable to fully relax contraction in porcine-skinned fibers in the absence of Ca2+. p.Arg145Trp mutation also led to an increase in the Ca2+ sensitivity of force development compared with wild type cTnI.9)
The clinical features of this family are interesting for several reasons. First, it is well-known that the p.Arg145Trp mutation in the TNNI3 gene could induce either HCMP or RCMP in a single family. The mixed appearance of HCMP and RCMP had previously only been reported in a case with the p.Asp190His mutation (not the p.Arg145Trp mutation) in the TNNI3 gene.6) The second important observation is that the majority of the individuals carrying the p.Arg145Trp mutation showed a near-normal phenotype of the heart at the time of presentation. In particular, the proband's mother (II:3) was healthy without any signs of CMP, except for subtle changes in her ECG and echocardiogram, even at the advanced age of 81. Therefore, the prognosis of RCMP with p.Arg145Trp mutation could be more favorable than that of other TNNI3 mutations. RCMP is a rare and distinct form of CMP that is characterized by diastolic dysfunction but intact systolic function, until the later stages of the disease.1) Among the different CMPs, RCMP has the worst prognosis.14)15) According to published results,6) RCMP patients with the p.Arg145Trp mutation showed a later onset of the disease in comparison with those having other TNNI3 mutations, including p.Arg192His, p.Lys178Glu, p.Ala171Thr, and p.Leu144Gln (median age, 69 years vs. 25 years). In our cases, one affected individual (III:2) died at 58 years, whereas other individuals who harbored the TNNI3 p.Arg154Trp mutation were all asymptomatic at the time of presentation, suggesting phenotypic diversity exists in prognosis as well as subtypes of CMPs.
It is still unclear why the same mutation causes different CMP phenotypes. It has been suggested that different mutations produce different sensitivities to Ca2+; high sensitivity leads to HCMP or RCMP, and low sensitivity to DCMP.4) However, the lack of a molecular understanding of the TNNI3 mutations and their functional consequences has limited our ability to address how phenotypic diversity arises from the same gene mutation. It is possible that even in families with Mendelian inheritance of CMP, more complex genetic or environmental interactions are involved in determining the clinical phenotype.4) In fact, it has been reported that 9.5% of Chinese HCMP patients harbor multiple mutations of sarcomere proteins, and the number of mutations was positively correlated with the maximum wall thickness.16) Therefore, we could not exclude the possibility of the presence of another gene mutation in patients with severe phenotypes. Also, it is highly speculative that the non-appearance of the clinical expression of CMP in the mother may be the consequence of the opposed influence explicated by another hypothetical gene sequence having a protective effect.
In conclusion, we report a large family with the TNNI3 p.Arg145Trp mutation and a diverse range of phenotypes from RCMP to HCMP to near-normal heart. To our knowledge, this is the first genetically confirmed CMP case with diverse clinical expression in the Korean population. An interesting question for future research will involve determining how mutations in the same gene can cause a range of distinct phenotypes.


 XML Download
XML Download