

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Atherosclerosis, a disease of blood vessel walls characterized by thickening of the artery walls,1)2) is a chronic inflammatory disease in which immunological pathways play essential roles.3)4) Atherosclerotic plaques contain immune cells, vascular cells (i.e. endothelial cells and vascular smooth muscle cells), extracellular matrix, cellular debris, and lipids. During the initial stage of plaque formation, lipids accumulate in the intima, where these lipids, such as low-density lipoprotein (LDL), are modified by oxidation or by enzymes to form oxidized LDL (oxLDL), and a chronic inflammatory response develops to these modified lipids. Lipid-laden macrophages known as foam cells are the most prevalent cells in early atherosclerotic plaque lesions (fatty streaks), although these plaques also contain T cells. Inflammatory cytokines produced by the accumulated immune cells, which include T cells and antigen presenting cells (APCs), such as macrophages and dendritic cells (DC) that present antigenic peptides including oxLDL, affect the development of atherosclerosis.5)6)7) Over time, fatty streaks can progress into mature, advanced atherosclerotic plaques. Mature plaques generally have abundant accumulation of macrophages and T cells. These immune cells are activated and produce high levels of inflammatory cytokines such as INF-γ and tumor necrosis factor (TNF). When these plaques progress into more complex lesions, the lipid core region becomes a large necrotic core attributing to the death of macrophages and vascular smooth muscle cells (VSMCs)8)9) and elevated levels of matrix metalloproteinases (MMPs), which degrade the extracellular matrix, leading to weakened fibrous caps.10)11) Moreover, mature plaques are more prone to rupture, causing atherothrombotic vascular conditions such as a myocardial infarction. However, there are presently no established atherosclerotic plaque rupture murine models.
CD137 is a member of tumor necrosis factor receptor superfamily (TNFRSF), and its alternative names are TNFRSF9, 4-1BB, and induced by lymphocyte activation (ILA). CD137 was originally discovered on activated T cells, so the best characterized function of CD137 is its co-stimulatory activity for activated T cells. Interaction of CD137 and its ligand CD137L enhances T cell proliferation, effector function, and survival. Among the various animal disease models, CD137 was the most investigated target molecule in tumor models.12) It has been reported that CD137 is expressed on a wide range of tumor cells,13) and both in tumor vessel walls and tumor-liver tissue in patients with cancer.14)15) Therefore, many investigators have studied the anti-tumor effects of CD137 as a therapeutic agent.
Accordingly, recent studies referred to CD137 and its ligand CD137L signaling as a potential biomarker between the immune system and cardiovascular disease including atherosclerosis. This signaling could be an important therapeutic target for development and plaque stability of atherosclerosis. Here, we discuss the characteristics of CD137 in immune and vascular cells, and its role as the diagnosis biomarker for the development of atherosclerosis.
T cells in atherosclerosis
T helper (Th) cells
T cells are present at all developmental stages of atherosclerotic plaques (~10% of plaques); they and their cytokines play important roles in disease processes.16)17) The majority of T cells in mouse and human atherosclerotic plaque are CD4+ T cells that express the αβ T cell receptor (TCR) and have an effector T cell phenotype. With respect to cytokine profiles, effector T cells are functionally classified as T helper cell type 1 (Th1), Th2, or Th17. It has been demonstrated that the Th1 response in atherosclerosis shows predominantly pro-atherogenic potential. Th1 cells are characterized by their prominent production of pro-inflammatory cytokines, including interferon (IFN)-γ, tumor necrosis factor (TNF)-α, interleukin-2 (IL-2), and IL-7, which activate macrophages as well as other immune cells. The interaction of the T cell receptor (TCR) on activated Th1 cells with major histocompatibility complex II (MHC II)-binding antigen peptides delivered by APCs synergistically induces the bidirectional activation of both cell types.18)19)20) T cell activation is further enhanced by ligand binding to co-stimulatory receptors including CD40, CD137, and OX40, which are expressed on the cell surface. Enhanced T cell activation by ligand binding to co-stimulatory receptors significantly augments cytokine release by monocytes/macrophages, further exacerbating inflammation and promoting atherosclerosis.21)22) Th2 cells produce Th2-related cytokines, including IL-4, IL-5, and IL-13, which are known to antagonize Th1 effects and thereby produce atheroprotective effects.23)24) However, the role of the Th2 response during the development of atherosclerosis remains uncertain, seemingly depending on the stage of the atherosclerotic lesion formation or the experimental mice model employed. Recently, there has been research focused on IL-17-producing Th17 cells, which play important roles in the pathogenesis of atherosclerosis. Studies on atherosclerosis have led to conflicting results regarding the role of Th17 cells in disease development. IL17-A and IL-17A expressing T cells (Th17) were significantly increased in ApoE-/- mice compared with age-matched C57BL/6 mice. When feeding them a high fat diet, Th17 cells were elevated in the aorta. Blockade of IL-17A in ApoE-/- mice by use of adenovirus-produced IL-17 receptor A (IL-17RA) reduced the formation of atherosclerotic plaque. In the study using genetically deficient IL-17A and IL-17 RA in ApoE-/- mice, IL-17A/IL-17RA axis increase aortic arch inflammation through the induction of aortic chemokines and leukocytes recruitment during atherogenesis.25)26) In contrast, several studies suggested that IL-17 production protects against vascular inflammation and plaque formation in Ldlr-/- mice, and supplementation with IL-17 reduces vascular inflammation and limits development of atherosclerotic plaque. And IL-17A mainly produced by Th17 cells plays a antiatherogenic role in a myocardial injury.27)28)
Regulatory T (Treg) cells
As atherosclerosis is a chronic inflammatory disease, and Th1-type cytokines dominate in mouse and human atherosclerotic plaques, immune homeostasis is crucial for protection and stabilization of atherosclerotic plaques. Two anti-inflammatory cytokines, interleukin-10 (IL-10) and transforming growth factor-β (TGF-β), are counterbalancing cytokines that dampen disease activity. Several cell types can produce IL-10 and TGF-β, including platelets, M2 macrophages, endothelial cells, vascular smooth muscle cells, and regulatory T (Treg) cells. CD4+CD25+ Treg cells constitute 5-10% of peripheral CD4+ T cells, which actively maintain an immunological tolerance to self-antigens, and control the development and progression of atherosclerosis.29)30)31) Treg cells actively suppress activation or effector functions of Teff cells, either directly or through effects on APCs. Foxp3, an X-linked transcription factor that is highly and specifically expressed in Treg cells, is a lineage specification factor and has a critical role in Treg suppressive function.32)33) CD4+CD25+ Treg cells are produced in the thymus as a functionally mature subpopulation of T cells (natural Treg, nTreg) and can also be induced from naïve T cells in the periphery (adaptive Treg). Deficiencies in the development or function of these cells are associated with severe autoimmune inflammatory diseases. Many studies have suggested that Treg cells are important regulators in the development of atherosclerosis.34) Many studies have suggested that Treg cells were found in both mouse and human atherosclerotic plaques and showed that Treg numbers are reduced during atherosclerosis. In a mouse study, ApoE-/- mice have reduced numbers of Treg cells compared with C57BL/6 mice,35) and feeding with a high fat diet also induced a reduction in Treg cells compared with mice fed a regular normal chow diet.36) Also, deficiency of Treg cells has been reported to increase atherosclerosis and plaque inflammation,37)38) and the anti-inflammatory cytokines produced from Treg cells, IL-10 and TGF-β, have been shown to attenuate the development of atherosclerosis.39) In many studies of Treg cells functioning during this disease, Treg cells showed the ability to inhibit both IFN-γ producing Th1 and IL-17 producing Th17 subsests of Teff cells.40) Treg cells can also inhibit pro-atherogenic macrophage inflammation by promoting the differentiation of M1 macrophages towards anti-inflammatory M2 macrophages. Therefore, expansion of Treg cells enhanced atherosclerotic plaque stability by inducing collagen producing M2 macrophages. Treg cells reduces the transition of macrophages into foam cells by inhibiting lipid accumulation via down-regulation of scavenger receptor class A (SR-A), and CD36.41) In addition, overexpression of IL-10 reduces VLDL, LDL levels in Ldlr-/- mice,34) and adaptive transfer of Treg cells also inhibit endothelial activation, leading to reduction of leukocytes attachment.42)
Co-stimulatory molecules and receptors
As stated above, T cells are initially activated by the interaction of TCR and MHC-binding antigen peptides delivered by APCs to proliferate and differentiate into Teff cells. In contrast to this antigen-specific first signal, the second signal for T cell activation is antigen-non specific and generated by co-stimulatory molecules. At each stage, T cells are activated by the combination of these two signals, both delivered by APCs. Dendritic cells (DCs), major member of the APCs, can initiate activation of T cells by presenting antigens into MHC-binding peptides.43)44) Co-stimulatory pathways are necessary for T cell activation; many studies have suggested that their absence can lead to functional inactivation of T cells, and cause T cell death by apoptosis. Therefore, these co-stimulatory signals play a critical role in the induction of an adaptive response, and provides a mechanism to communicate immunological danger to the adaptive immune system.45)46) Both APCs and adaptive immune cells (T and B cells) respond to receptor signaling generated by co-stimulatory ligands. B cells also require two types of signals to become activated and produce antibodies. B cell binds antigens with its BCR (B cell receptor, a membrane-bound antibody) as a first signal. And activated T cells generally provide the second signal for B cell activation through co-stimulatory secondary signals, leading to mount a humoral response.47) The interaction between T cells and B cells mediated by this signaling pathway is responsible for B cell proliferation and differentiation, immunoglobulin isotype switching, and antibody secretion. There are two families of costimulatory molecules, namely the B7 family and the tumor necrosis factor superfamily (TNFSF). Several members of the TNFRSF, including CD40, LIGHT, CD134 (OX40), and CD137, have been shown to carry out important immune modulatory functions in atherosclerosis.48)49)50)51)
CD137 and CD137L
CD137 is a 30 kDa type I transmembrane glycoprotein, and is a well-known T cell co-stimulatory molecule. Expression of CD137 is activation dependent. CD137 is not detected (<3%) on resting T cells or naïve T cells. However, CD137 is upregulated when T cells are activated upon stimulation. CD137 has been reported to be expressed on most immune cells, including activated CD4+ and CD8+ T cells, natural killer (NK) cells, NKT cells, and CD4+CD25+ Treg cells. CD137 is also expressed on myeloid cells, such as monocytes, neutrophils, mast cells, eosinophils, and dendritic cells.52)53)54) In vascular cells such as endothelial cells and vascular smooth muscle cells, CD137 is expressed in an activation-dependent manner. CD137L, a ligand for CD137, is a type II membrane glycoprotein of the TNF superfamily that is expressed on APCs such as monocytes, macrophages, dendritic cells, and activated B cells. In general, TNFRSF members are initiators of inflammatory diseases. CD137-deficient mice have been used in an attempt to define the role of CD137 in the immune system and in immune-related diseases. CD137-deficient mice have been shown to exhibit an enhanced T cell response, but have normal T cell development.55)56) Vinay and colleagues also showed that CD137-deficient mice have a reduced number of natural killer cells and natural killer T cells, which leads to a resistance to lipopolysaccharide-induced shock syndrome.57) In a dendritic cell study, CD137 was found to be a crucial survival factor58) and to control certain regulatory activities by promoting production of retinal dehydrogenase.59) Therefore, CD137 appears to play a variety of roles in the immune system.
CD137 and CD137L in atherosclerosis
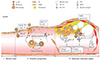
With regard to the formation of atherosclerotic plaque lesions, a few studies have reported that CD137 and CD137L play critical roles in this process. For example, Olofsson and colleagues observed clear CD137 expression in human atherosclerotic plaques.50) They also showed that CD137 was expressed on T cells and endothelial cells in human atherosclerotic plaque lesions. CD137 is inducible in vascular cells, including endothelial cells and vascular smooth muscle cells, in vitro. In clinical studies, CD137 expression is known to be involved in the stability of coronary atherosclerotic plaques.60)61) Patients with ACS (acute coronary syndromes) showed both increased levels of CD137 expression in monocytes and soluble CD137 in serum compared with control and SA (stable angina) group, deriving a conclusion of positive relationship between CD137 expression and increase in serum MMP-3, and MMP-9 in patients with ACS. In addition, Patients with ACS showed an increase in both soluble and membrane-bound CD137 expression, leading to a positive correlation with the CRP level in patients with ACS. Therefore, we speculate that CD137 could be a typical marker for instability of atherosclerotic plaques in patients with ACS. CD137-CD137L interaction induces activation of the phospholipase C (PLC) signaling a pathway in human umbilical vein endothelial cells (HUVECs).62) Both plasma-soluble CD137L and CD137L expression on monocytes were upregulated in patients with atherothrombotic stroke63) and acute coronary syndrome.64) Based on results of a study using an atherosclerotic mouse model, it has been suggested that CD137-CD137L interaction regulates atherosclerosis via cyclophilin A.65) Our group previously showed that CD137 deficiency reduced the formation of atherosclerotic plaque lesions in a mouse atherosclerosis model.51) Briefly, we found the molecular mechanisms whereby CD137-CD137L interaction induces activation of macrophages and Teff cells through bidirectional signaling. Activated Teff cells produce more IFN-γ, which leads to recruitment and activation of macrophages. The activated macrophages produce more TNF-α and MCP-1, which cause endothelial CD137 expression. This endothelial CD137 signaling induces the production of MCP-1 and cell adhesion molecules, probably leading to enhancement of leukocyte recruitment to the lesion (Fig. 1).
Advanced, vulnerable plaque
During the progression of atherosclerosis, lipids and leukocytes accumulate in the intima, causing activation of the immune system. Inflammatory cytokines produced by the accumulated immune cells, including T cells and APCs, affect the development of atherosclerosis. Over time, mature plaques develop into vulnerable plaques, which are more prone to rupture, causing subsequent atherothrombotic vascular conditions such as myocardial infarction. Therefore, lesion growth in advanced plaques differs significantly from early lesions.
Pathological features of vulnerable plaques
Vulnerable plaques generally have a large necrotic core, which is attributed to the death of macrophages and vascular smooth muscle cells (VSMCs). Ruptured plaques are characterized by a necrotic core with an overlying thin fibrous cap infiltrated by macrophages and lymphocytes. The extracellular matrix, which includes collagen fibers, normally confers biochemical stability to the fibrous cap of the atherosclerotic plaque. Vulnerable plaques have elevated levels of matrix metalloproteinases (MMPs), which may degrade the extracellular matrix, leading to weakened fibrous caps. These proteolytic enzymes released from macrophages in atherosclerotic plaques exacerbate matrix degradation, leading to lesion instability.66) Previous studies in a mouse model of atherosclerosis suggested that levels of MMP-2 and MMP-9 increase during atherosclerosis, and that MMP-2 and MMP-9 expression largely depends on bone marrow-derived macrophages.67)68)69) Furthermore, it has been suggested that over expression of MMP-2 and MMP-9 in macrophages induces vulnerable features of plaques.70)71) Cathepsins, a subset of cysteine proteinases, have pivotal roles during atherosclerosis. Previous mouse studies have suggested that cathepsin L stimulates autophagy and inhibits endothelial cell apoptosis.72) Deficiency in either cathepsin L or cathepsin S reduces the development of atherosclerosis.73)74) Leukocyte cathepsin K is an important factor in atherosclerotic plaque vulnerability and vascular remodeling.75)76)77) It has been suggested that the pro-inflammatory cytokine IFN-γ secreted from activated T cells (Th1) inhibits the synthesis of new collagens by VSMCs in advanced vulnerable plaques, and also inhibit the proliferation and expression of smooth muscle alpha actin of VSMCs.78)79) In this pro-inflammatory condition, VSMCs could be main targets for pro-inflammatory mediators, and growth factors like platelet-derived growth factor (PDGF) as well as injured vascular endothelial cells (ECs) and VSMCs themselves, leading to quiescent healthy VSMCs into a proliferative synthetic form, a so called "phenotype change".80)81)
TNFSF/TNFRSF and vulnerable plaques
A recent study suggested that ligation of CD40 (TNFRSF5) expressed on macrophages to CD40L (TNFSF5) expressed on T cells causes overproduction of matrix-degrading proteases, including MMP-1, 8, and 13, that induce collagen breakdown.82) A recent mouse study found that CD40 is involved in atherosclerotic plaque stability.48) Deficiency of CD40 reduced atherosclerosis and led to a stable atherosclerotic plaque phenotype by inducing plaque fibrosis and the M2 macrophage phenotype. However, deficiency of CD40 did not affect apoptosis of cells in atherosclerotic plaque lesions. In addition, OX40 (TNFRSF4) and its ligand OX40L (TNFSF4) in CD4 T+ lymphocytes were highly upregulated,83) and both OX40L on platelets and soluble OX40L in serum were increased in patients with acute coronary syndrome.84) It has been suggested that LIGHT (TNFSF14) and LIGHTR (LIGHT receptor, TNFRSF14) are expressed in atherosclerotic plaques, and decrease plaque stability by inducing the expression of TNF-α, IL-8, and MMPs.85) Furthermore, activation of TNFRSF12 induces MMP expression, leading to destabilization of atherosclerotic plaques.86)
CD137 and vulnerable plaques
Olofsson and colleagues50) suggested that CD137 was induced in VSMCs by proinflammatory cytokines in atherosclerotic plaques, and that stimulation of these VSMCs with CD137L reduced proliferation. These results indicated that CD137 signaling may contribute to the destabilization of atherosclerotic plaques. Recently, our group determined the CD137-mediated molecular mechanisms whereby activation of CD137 signaling decreases the stability of advanced atherosclerotic plaques via its combined effects on Teff cells, VSMCs, and macrophages.87) Briefly, CD137 increases the infiltration of Teff cells into plaque lesion sites, resulting in increased IFN-γ expression. Teff cell-derived IFN-γ then inhibits collagen synthesis in atherosclerotic plaques. Furthermore, CD137 activation leads to increased apoptosis of VSMCs, possibly by decreasing the antiapoptotic regulator, Bcl-2, and subsequently upregulating a cleaved caspase-3. In macrophages, activation of CD137 signaling boosts the oxidized low-density lipoprotein-induced expression of MMP-9 via the p38 mitogen-activated protein kinase and extracellular signal-regulated kinase 1/2 signaling pathways (Fig. 1).
Future directions
Many human and mouse studies have indicated that CD137 is a pivotal atherosclerosis-promoting factor. It is not surprising that CD137 functions not only in T cells but also in most immune and vascular cells. Recent studies have begun to address the roles of different DC subtypes in mouse atherosclerosis. Choi and colleagues evaluated DC populations in the mouse aorta. They showed that there are two DC subsets, CD11b+F4/80+CD14+DCSIGN+ monocyte-derived DCs (Mo-DCs) and Flt3-dependent CD11b-F4/80-CD103+Langerin+ classical DCs (cDCs), in the normal mouse aorta. They also found that Flt3-dependent cDCs play a protective role in the development of atherosclerosis by regulating the homeostasis of Treg cells, via studies using Flt3-/-Ldlr-/- mice.88) CD137 signaling has been extensively studied in both DCs and Treg cells. It has been suggested that CD137 is expressed on follicular DCs (FDC) in germinal centers, and mediates the activation of B lymphocytes.89) CD137 increases DC survival, and promotes both antigen-specific CD4+ T cell responses90) and the production of inflammatory cytokines.91) Studies on the gut mucosa have revealed that CD137 regulates retinal dehydrogenase (RALDH) expression and activity in DCs.92) Although there have been numerous studies investigating the functional effects of CD137 on DCs and Treg cells, limited information is available on the precise roles of CD137-mediated functions of specific subsets of DCs that regulate the homeostasis of Treg cells in a steady state and during the development of atherosclerosis. Future studies may elucidate the mechanisms of cross-talk between DC subpopulations and Treg cells, which constitutes a promising therapeutic target (Fig. 2).

 XML Download
XML Download