

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia and leads to increased risks of ischemic stroke, heart failure, and mortality. The socioeconomic burden of AF continues to increase; in the United States, the annual healthcare cost associated with AF is more than 6.5 billion dollars.1) Electrical, contractile, and structural remodeling are important contributors to AF genesis, and fibrosis is the hallmark of structural remodeling in the heart.2)3) Increasing evidence suggests that atrial fibrosis plays an important role in thrombo-embolic stroke in addition to being a substrate for AF, even though the various components that cause a prothrombotic and hypercoagulable state might primarily be involved in the pathophysiologic mechanisms of thrombo-embolic events in AF.4)5) Upstream therapy involving the renin-angiotensin-aldosterone system (RAAS), such as angiotensin-converting enzyme (ACE) inhibitors or angiotensin-II type 1 receptor blockers (ARBs), can reduce atrial stretch and fibrosis, and the reverse remodeling process by RAAS inhibition might decrease the development of AF.6)7) However, it remains largely unknown whether RAAS inhibitors directly affect thrombogenecity, preventing thrombo-embolism in AF. We aimed to determine whether ARBs can reduce thrombogenicity in tachy-induced AF canine models systemically or through reverse-remodeling of the atrium.
Materials and Methods
Study materials
All animal experiments in this study were performed according to procedures approved by the Korea University Institutional Animal Care and Use Committee. Thirteen dogs (20-30 kg) were used in this experiment. During pacemaker implantation, all dogs were anesthetized using xylazine (Rompun, 0.05-0.15 cc/kg I.M.; Bayer Korea, Ansan, Korea) and zolazepam (Zoletil 50, 2.5-7.5 mg/kg I.M.; Virbac, Carros, France), and general anesthesia was maintained using mechanical ventilation with intubation under halothane. After right thoracostomy, pacemaker leads were implanted into the epicardial wall of the right atrial appendage or right atrium (RA) and were connected to a generator (Medtronic Co. Ltd., Minneapolis, MN, USA) in the subcutaneous pocket (Fig. 1A). After the chest wall was surgically closed, postoperative care including administration of antibiotics was performed.
Fig. 1
The procedure under thoracostomy and electrograms during device interrogation. (A) Implantation of pacemaker with a generator and a lead for rapid atrial pacing. (B) Sinus rhythm before pacing. (C) Rapid pacing in the right atrial appendage. (D) Atrial fibrillation was identified after four weeks of pacing. RAA: right atrial appendage.

Study design
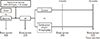
The overall design of this study is shown in Fig. 2. Surface electrocardiography (ECG) was monitored to identify sinus rhythm before pacing (Fig. 1B). One week post-surgery, after we confirmed that the general condition of each dog was good and the suture site was clean, rapid atrial pacing was continuously delivered over 4-8 weeks until sustained AF was induced (Fig. 1C and D). Rapid atrial pacing was initiated at a rate of 400 beats/min using 2 ms pulses at twice the threshold current and continued for 4 weeks. The presence of induced AF was identified on the electrogram stored by the pacemaker device, and ECGs were checked every one week to ascertain whether AF was sustained. The pacing rate was increased to 510 beats/min (maximal pacing rate of the device) when AF was not shown on the follow-up 12-lead surface ECGs. If sustained AF developed, the high-rate pacing was discontinued. During device interrogation and ECGs, anesthesia was applied using the same drugs as above. Candesartan cilexitil 10 mg/kg/day (AstraZeneca, Mölndal, Sweden), which was prepared at 1 mg/mL suspended in a 5% gum arabic solution, was orally administered for 12 weeks.
Biochemical measurement
Both arterial and venous blood was obtained via the femoral artery and vein at baseline and 0, 4, and 12 weeks after AF onset in the control and candesartan groups. All dogs were in AF at the time of blood sampling. Citrated whole blood from each sampling was immediately centrifuged and stored at -80°C before analysis. The serum levels of tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) for fibrosis, von Willebrand factor (vWF) for endothelial dysfunction, P-selectin for platelet activation, and vascular cell adhesion molecule-1 (VCAM-1) for an adhesion molecule were quantitatively measured using a canine-specific enzyme-linked immunosorbent assay kit (ElAab, Wuhan, China).
Histologic examination
Hearts were removed at the end of the experiments. Tissue specimens of both atria and their appendages were obtained and fixed in formalin solution for 24 hours. Each endocardial and myocardial section was cut into slices. Hematoxylin-eosin and Masson's trichrome were used to stain deparaffinized sections. Endocardial and myocardial fibrosis was semi-quantitatively assessed in whole tissues, excluding subendocardial or perivascular areas with a high amount of fibrosis. The amount of fibrosis in the whole tissue was graded using a semi-quantitative scoring system (0=absent, 1=mild, 2=moderate, & 3=severe fibrosis).8) A cardiovascular pathologist performed the analyses in a blinded manner.
Statistical analysis
Data analyses were performed using SPSS Statistics 17.0 software (SPSS Inc., Armonk, NY, USA). All values are expressed as mean±standard error of mean. Continuous values were compared with a paired t test in order to evaluate differences between time intervals. Categorical values were analyzed using the Kruskal-Wallis test. Statistical significance of serial changes in bio-markers was determined by analysis of variance (ANOVA) with repeated measures ANOVA. p≤0.05 were considered statistically significant.
Results
Canine atrial fibrillation models
Tachy-paced AF was induced in nine canines. One of the nine dogs died one month after AF onset from significant bradycardia during anesthesia. After AF was induced by pacing, sustained AF was confirmed in the remaining eight dogs with both pacemaker interrogation and 12-lead surface ECGs throughout the study period. A total of 12 dogs were randomly assigned to either the control group in AF (n=4), the candesartan group in AF (n=4), or the sham group (n=4). Table 1 shows baseline characteristics of the study subjects. There were no significant differences in age, sex, or body weight among the three groups, and there were also no significant differences in age (18.0±6.9 vs. 16.5±5.2 years old, p=1.000), body weight (27.7±3.2 vs. 27.6±4.1 kg, p=0.886), and male sex (50 vs. 50%, p=1.000) between the control group and the candesartan group.
Table 1
Baseline characteristics of study subjects

Arterial and venous bio-markers
At baseline, there were no significant differences in arterial or venous levels of TIMP-1, vWF, P-selectin, and VCAM-1 among the three groups (sham group vs. control group vs. candesartan group [Supplementary Table in the online-only Data Supplement]). There were also no significant differences in the arterial and venous levels of any bio-marker between the control group and the candesartan group. Fig. 3 shows the serum bio-marker levels according to time sequences after AF onset. There were no significant differences in arterial or venous levels of TIMP-1, vWF, P-selectin, or VCAM-1 between the control group and the candesartan group at AF onset. There were also no significant differences in the serum levels of TIMP-1, vWF, or P-selectin at either 4 or 12 weeks after AF onset between the two groups. Venous levels of VCAM-1 showed no significant change between the two groups from 4 to 12 weeks. Thus, there were no significant changes in the serum levels of bio-makers in the control and candesartan groups. Arterial VCAM-1 tended to decrease in the candesartan group from 4 to 12 weeks compared to the control group, although the difference was not statistically significant (301.0±38.6 to 160.1±70.4 in the candesartan group vs. 258.7±75.2 to 237.6±72.2 ng/mL in the control group, p=n.s.). Repeated measures ANOVA p-values for testing overall mean levels of bio-markers between the control group and the candesartan group did not significantly change across time periods.

Endocardial and myocardial fibrosis
Table 2 shows the fibrosis grades in the atrial endocardium and myocardium obtained from heart specimens in each group. The grades of endocardial fibrosis but not myocardial fibrosis after 12 weeks seemed to be reduced in the candesartan group compared to the control group. However, there was no statistically significant difference in either endocardial or myocardial fibrosis grade among the three groups, and there was also no significant difference in fibrosis grades between the control group and the candesartan group. Fig. 4 shows representative histological sections from each group.
Table 2
Fibrosis grades of study subjects by group

Fig. 4
Representative figures of atrial fibrosis in endocardium and myocardium. Subendocardial fibrosis was evaluated with Masson-Trichrome (MT) stain, in which bright blue-colored collagen bundles were easily identified. (A) Endocardial fibrosis in sham group. Subendocardial collagen deposition was marked in the control group (B) and mild in the candesartan group (C). Myocardial fibrosis was also evaluated on MT stain. (D) Myocardiual fibrosis in sham group. Intermyositic collagen deposition was slightly increased in the control (E) and candesartan groups (F). However, there was no statistically significant difference. Original magnification×200.

Discussion
Principle findings
The main findings of this study are as follows. First, the serum levels of bio-markers obtained from either the artery or the vein were not significantly different between the control group and the candesartan group during AF. Second, there were no significant changes in overall mean levels of the bio-markers between the two groups across the time periods studied. There was a trend towards decreased arterial VCAM-1 from 4 to 12 weeks in the candesartan group compared to the control group. Third, there was no significant difference in the grades of endocardial or myocardial fibrosis in either atrium between the two groups although endocardial fibrosis 12 weeks after AF onset seemed to be reduced in the candesartan group compared to the control group.
Angiotensin-II receptor blocker for fibrosis in atrial fibrillation
AF presents with both structural and electrophysiological abnormalities that are caused by diverse pathophysiological mechanisms. The remodeling process leads to atrial fibrosis, which is thought to be one of the critical components in generating arrhythmogenic substrates for AF.9)10)
RAAS has been shown to be involved in structural remodeling and electrophysiological abnormalities in the cardiac myocardium, leading to an increase in susceptibility to arrhythmia and the development of AF.11)12) In addition to hemodynamic changes, derangements of RAAS activate multiple cell signaling cascades, promoting cardiac fibrosis.13) Among the components of RAAS, ACE and angiotensin II are known to substantially contribute to atrial fibrotic remodeling during AF; many studies have demonstrated the effect of RAAS inhibition on AF. ACE inhibitors interfere with signal transduction targeted to angiotensin II in congestive heart failure.14) Kumagai et al.7) demonstrated the reverse-remodeling effect of ARB that mitigated atrial fibrosis and development of AF. At the cellular level, rapid atrial pacing induced the release of angiotensin II, leading to activation of the angiotensin II type 1 receptor and causing a downregulation of Smad7 in the atria and a decrease in inhibitory feedback of TGF-β1.15) Kim et al.16) reported that losartan, an ARB, disrupts collagen fiber formation and prevents the alteration of tissue nitric oxide synthase levels, which may prevent subsequent AF induction. Park et al.17) demonstrated that the left atrium (LA) had a more advanced matrix and subendocardial remodeling compared to the RA in patients with mitral valvular AF; however, expressions of tissue factors associated with thrombogenesis were not significantly different between the RA and the LA. Unlike previous reports, we assessed the degrees of both endocardial and myocardial fibrosis in this study. Our data showed that endocardial fibrosis in LA but not myocardial fibrosis was reduced by ARB, although the change was not significant. This finding assumes that, unlike the direct effect on the endocardium, perfusion or delivery of ARBs to the myocardium are affected by pacing-induced heart failure. In addition, the effect of ARBs seemed to be less beneficial in RA. However, further investigation with a longer period of treatment is required to compare the differential effects on the RA and LA.
Angiotensin-II receptor blocker for thrombogenesis in atrial fibrillation
There is a paucity of information regarding the direct effect of ACE inhibitors or ARBs on thrombogenicity in AF. Thus, we measured both atrial fibrosis and the serum levels of four bio-markers, TIMP-1 for fibrosis, vWF for endothelial dysfunction, P-selectin for platelet activation, and VCAM-1 for an adhesion molecule, in order to determine whether ARBs inhibit thrombogenicity in tachy-induced AF canine models systemically or through reverse-remodeling of the atrium.
It is well known that AF is associated with a higher risk of stroke and thromboembolism, but the mechanism underlying thrombogenesis in AF is complex. Increasing evidence supports the idea that many components are involved in the prothrombotic or hypercoagulable state in AF, including abnormalities in vessel walls, flow, and blood constituents.5) RAAS may play a role in the mechanisms of prothrombosis in AF. Upregulation of angiotensin II in AF has been demonstrated to have various proinflammatory properties such as increased cytokines and chemokines; it has also been shown to activate matrix metalloproteinase and thromboxane A2, leading to prothrombogenesis in AF.18)19) Based on this modulation of the RAAS cascade, ARBs are a potential therapeutic target for reducing thrombosis and stroke. A study in patients with AF of the Losartan Intervention For Endpoint reduction in hypertension trial showed that losartan is more effective than atenolol-based therapy in reducing the risk of the primary composite endpoint of cardiovascular morbidity, mortality, and stroke;20) however, whether the RAAS blockade directly influences thrombogenesis or its effect is secondary to reverse remodeling of AF substrates remains unknown.
Goette et al.4) reported that rapid pacing of atrial tissue slices increased VCAM-1 expression, which was abolished by pretreatment with olmesartan, and alterations of in vivo VCAM-1 expression were also attenuated by infusion of irbesartan. In their study, ARBs were administered via direct infusion over a short time. To the best of our knowledge, our study is the first to investigate the effect of long-term administration of ARBs on prothrombogenic processes in pacing-induced AF canine models. Our results showed that the level of arterial VCAM-1 increased 4 weeks after AF onset in both the control and candesartan groups, and, after that, decreased only in the candesartan group. The effect of ARB on arterial VCAM-1 seemed to be delayed. This finding suggests that arterial VCAM-1 is reduced by ARBs, probably through secondary effects of the reverse remodeling process as well as direct effects on the reduction of arterial VCAM-1.
Limitations
Our study was conducted using a small number of subjects, which may explain why our results were not statistically significant. In this study, candesartan cilexitil was orally administered at a dose of 10 mg/kg/day, which was the maximum tolerable dose that did not reduce blood pressure; thus, we did not measure blood pressure during the experiment. However, there is a possibility that ARBs decreased blood pressure in the study subjects; hypotension could have affected our results. Neither continuous telemetric ECG monitoring nor implantable loop recorder was done, but both pacemaker interrogation and 12-lead surface ECGs were used to determine the atrial rhythm and evaluate whether or not AF was sustained. We did not use quantitative methods for assessing the effects of fibrosis. Histologic results were likely to have a ceiling effect because the grading of fibrosis was semi-quantitatively assessed. In this study, the degree of fibrosis was measured using a semi-quantitative scoring method because the density of histological stain varies when tissue thickness varies. In addition, quantitative methods may be biased in such a small sample size. Although we did not measure the data using quantitative methods, semi-quatitative methods have been validated.8) In order to minimize potential bias, a cardiovascular pathology specialist assessed the extent of fibrosis in a blinded manner. Western blotting for protein expression in tissue specimens was not performed.
Conclusions
In conclusion, this study did not show that candesartan significantly affected thrombogenicity or fibrosis during AF. Candesartan seemed to decrease the level of the arterial adhesion molecule VCAM-1 and endothelial fibrosis in this study, suggesting that RAAS may have a pathophysiological role in the prothrombogenic process and that ARB is a potential treatment for thrombogenicity and endocardial remodeling in AF. Further experiments using a larger number of subjects and future studies in the clinical setting are warranted to determine the therapeutic effect of RAAS blockade on the prothrombogenic process in AF.
 XML Download
XML Download