

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Since the original description of neuromuscular disorders (NMDs), it was noted that cardiac structures could be affected alongside the myopathies.1) Among the neuropathies, cardiac involvement (CI) was first described in a patient suffering from amyloid neuropathy.2) The type, frequency, management, and outcome of CI among the various NMDs vary significantly between the different disorders of the muscles and nerves. The aim of the present review was to summarise and discuss previous and most recent findings concerning the type, prevalence, diagnosis, treatment, and outcome of CI in NMDs.
Method
Data for this review were identified by investigating the archives of MEDLINE, Current Contents, Springerlink, Wiley, EBSCO, Ovid, and Web of science by applying a sensitive search strategy using combinations of the following search terms: "myopathy", "muscle disease", "muscular dystrophy", "transmission", "myasthenia", "neuromuscular", "nerve", "neuropathy", "motor neuron disease", "plexopathy", "radiculitis", "anterior horn cell", in combination with "heart", "cardiac", "cardiologic", "myocardium", "cardiomyopathy", "noncompaction", "hypertrabeculation", "Takotsubo", "echocardiography", "electrocardiography", "cardiac magnetic resonance imaging", "late gadolinium enhancement", "heart failure", "left ventricular dysfunction", "right ventricular dysfunction", "arrhythmias", "ventricular tachycardia", and "sudden cardiac death". Further manual searches were conducted to identify other relevant articles from cross-references. Randomized (blinded or open label) clinical trials, longitudinal studies, case series, and case reports were considered. Hereditary as well as acquired NMDs were considered. Abstracts, meeting reports, and animal studies were not included. Only articles regarding humans and published in the English-speaking literature between 1966 and 2015 were considered for inclusion. Appropriate papers were studied and assessed for their applicability to be incorporated in the present review.
Results
Altogether, 224 papers met the inclusion criteria and were included in the review. The full text was available for 165 of these papers whereas only the abstracts were available for 59 papers.
Classification of cardiac involvement
CI was categorised according to various criteria. Firstly, CI was classified according to the anatomical structure affected (i.e. myocardium, conduction system, valves, coronary arteries, aortic root, endocardium, pericardium). Secondly, CI was classified according to the physiological function that was impaired (i.e. heart failure, valve stenosis/insufficiency, pulmonary hypertension, impaired autonomic innervation). Thirdly, CI was classified according to the abnormality found on further diagnostic work-up, such as systolic dysfunction, late gadolinium enhancement (LGE), mitral valve prolapse syndrome (MPS), pulmonary valve stenosis, concentric/eccentric myocardial thickening, arrhythmias, congenital anomalies.
Types of cardiac involvement
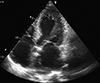
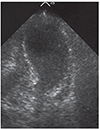
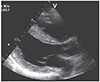
Cardiac involvement can manifest as hypertrophic cardiomyopathy (hCMP), dilated cardiomyopathy (dCMP), restrictive cardiomyopathy (rCMP), arrhythmogenic right ventricular dysplasia, histiocytoid cardiomyopathy (CMP), left ventricular hypertrabeculation/noncompaction (LVHT) (Fig. 1), or as Takotsubo syndrome (TTS) (Table 1 and Fig. 2). Involvement of the cardiac conduction system can present as spontaneous impulse generation or impulse conduction disorder. Valve disease in NMD may manifest as MPS whereas pathologic involvement of the coronary arteries can present with symptoms of coronary artery disease or coronary vascular spasms.3) NMD involvement of the aortic root may manifest as aortic root ectasia. Endocardial involvement may manifest as endocardial fibrosis exclusively affecting the endocardium or may also involve the myocardium (endocardial, subendocardial, mid-myocardial, trans-myocardial).4) Pericardial involvement by NMD can present as a pericardial effusion. Congenital abnormalities may affect any cardiac structure.
Neuromuscular disorders with cardiac involvement
Various NMDs can affect the heart and can develop before, during, or after pathologic involvement of the skeletal muscles and/or nerves. Furthermore, CI may dominate the clinical presentation or may be an insignificant part of the clinical presentation. Particularly, NMDs with multi-organ disorder syndrome, such as myotonic dystrophies or mitochondrial disorders (MIDs) may present with CI. NMDs with CI may be classified as disorders of the nerves with CI, transmission disorders with CI, or as myopathies with CI. CI is generally much more prevalent in muscle diseases as compared to neuropathies or transmission disorders.
Disorders of the peripheral nerves with cardiac involvement
Disorders of the nerves are classified as neuronopathies (also: anterior horn cell disorders), radiculopathies, plexopathies, or neuropathies.
Neuronopathies: CI in neuronopathies is uncommon but occasional case reports exist in the literature.5) CI in neuronopathies includes dilation of the left ventricle,5) dCMP,6) or arrhythmias7) in patients with spinal muscular atrophy (SMA). The case of a patient with a SMA phenotype due to a LMNA mutation who also developed arrhythmias and systolic dysfunction has been reported.8) CI in amyotrophic lateral sclerosis (ALS) can present with dCMP,9) cardiac sympathetic hyperactivity,10) arrhythmias,11) or TTS (Table 2).12) Admittedly, the diagnosis of ALS was not definitively determined in a subset of these cases. CI in bulbar spinal muscular atrophy (Kennedy disease) manifests as electrocardiographic (ECG)-abnormalities, which can be seen in up to half of these patients.13) CI in GM2-gangliosidosis (hexosaminidase deficiency, Sandhoff) manifests as MPS or mitral regurgitation.14) CI has not been described in adrenoleukodystrophy. However, further systematic, prospective studies on the CI in neuronopathies with a definitive diagnosis are warranted before the prevalence and management of CI in neuronopathies can be adequately assessed.
Radiculopathies: Conceivably, radiculopathies may cause cardiac disease, but in cases of radiculitis from infection with Borrelia burgdorferi or radiculopathy from amyloidosis, it is difficult to exonerate the infectious agent itself from directly causing cardiac disease as opposed to from amyloid deposition.
Plexopathy: Brachial plexus lesions from diabetes, viral infection, or trauma can theoretically affect autonomic cardiac function. However, no such reports were identified in the literature.
Neuropathies: CI in neuropathies has been described predominantly in the hereditary neuropathies and less commonly in the secondary neuropathies. Among the hereditary neuropathies, CI has been reported in hereditary transthyretin amyloidosis,15) hereditary sensory-motor neuropathy (HSMN), HSAN, Fabry disease, and Refsum disease (Table 2). CI in transthyretin amyloidosis is characterised by CMP,15) which may be independent of the neuropathy and due to primary deposition of amyloid in the myocardium, arrhythmias,16) rCMP, and heart failure. HMSN2 due to mutations in the DCAF8 can be variably accompanied by CMP.17) HSMN may also occur alongside TTS.18) Hereditary neuropathy due to a PMP22 duplication may be associated with dilatation of the ventricles, LVHT, or heart failure (Table 2).19)
Transmission disorders
CI has not comprehensively studied in patients with transmission disorders. In a study by Hofstad et al.,20) CI in myasthenia presented most commonly with arrhythmias. A small number of patients, particularly those with thymoma, can also develop myocarditis.20)21) In a study of 108 patients with myasthenia, 16% of these patients developed CI.20) In single cases, myasthenic crisis has been shown to trigger TTS.22)23)24)25)
Muscular dystrophies
Duchenne muscular dystrophy: In patients with Duchenne muscular dystrophy, cardiac disease occurs in nearly all cases if patients survive long enough and significantly determines the outcome. Cardiac disease in Duchenne muscular dystrophy (DMD) develops shortly after the onset of muscular manifestations. Initial manifestations of cardiac disease include abnormalities of impulse generation and conduction. Involvement of the myocardium usually becomes evident after patients have become wheelchair-bound. With disease progression, myocardial function, too, deteriorates and becomes a major outcome measure in these patients. CMP in DMD is characterised by progressive fibrosis of the myocardium, which can be most easily assessed by cardiac magnetic resonance imaging (cMRI).26)27) Progression of myocardial fibrosis is correlated with dilatation of the ventricles, decline of systolic function, and subsequent heart failure.28) A rare manifestation of CI in DMD is LVHT (Table 3).29) Various impulse generation and conduction abnormalities have also been reported in patients with DMD but it is unknown if the frequency of these abnormalities increases with worsening dCMP and progressive myocardial fibrosis. Patients may require implantation of a pacemaker because of third-degree AV-block. CI can be observed even in female carriers of the disease.30) Poor heart function in DMD is usually the cause of early death.31) Causes of sudden cardiac death (SCD) in DMD include sustained ventricular tachycardias, asystole, or acute heart failure.
Becker muscular dystrophy: Though BMD is due to mutations in the same gene as DMD and cardiac disease also occurs in BMD, there are several similarities and differences with respect to CI. As in DMD, the most frequent manifestations of CI are dCMP, cardiac conduction defects (CCDs), and arrhythmias (Table 3).32) Rarely, LVHT has been described in addition to dCMP in BMD patients.33) CI in BMD usually develops later during the course of the disease as in DMD.32) Only rarely may CI in BMD develop early in the natural history.34)35) In two siblings with BMD, dCMP was the initial manifestation of CI occurring at 11 years of age.34) One of the siblings died at 14 years while awaiting heart transplantation (HTX), whereas the other required implantation of a ventricular assist device.34) Few reliable data are available regarding the incidence of CI in BMD. CI in BMD is frequent, but most likely less frequent than in DMD.36) In a study of 48 BMD patients aged 26-56 years, 19% had dCMP.37) Given that patients usually die between their 40s and 80s, the prognosis is more favourable than it is with DMD patients.37)
Emery-Dreifuss muscular dystrophy: X-EDMD is due to mutations in the emerin or the FHL1 gene. CI in X-EDMD due to emerin mutations has been occasionally reported and manifests as CCDs.38) Rarely, dCMP in association with life-threatening ventricular arrhythmias has also been reported.38) A subset of patients with X-EDMD due to FHL1 mutations can present with pulmonary artery hypoplasia or SCD (Table 3).39) In another study, FHL1 patients presented with myocardial thickening, hCMP, and SCD.40) Single patients may also present with pulmonary valve stenosis.41) Autosomal dominant (AD)-EDMD is due to mutations in the LMNA gene and, like other laminopathies, frequently associated with cardiac disease. CI in AD-EDMD includes CCDs, dCMP, and heart failure.42) Rarely, hypoplasia of the aorta has been reported as a cardiac manifestation of AD-EDMD.43) Should atrial fibrillation develop in this patient population, ischemic stroke may be the initial manifestation of the muscle disease.44)
Limb girdle muscular dystrophies: Cardiac disease occurs in various types of LGMD but is particularly more common in the autosomal dominant LGMD1B laminopathy and the recessive LGMD types A (calpainopathy), B (dysferlinopathy), C (gamma-sarcoglycanopathy), D (alpha-sarcoglycanopathy), E (beta-sarcoglycanopathy), I (fukutin-related protein [FKRP]), and M (FKTN gene).45)46)47) Patients with LGMD1B may be severely affected by cardiac disease manifesting as CCDs, dCMP, or SCD.45) Some of these patients harboring duplications in the LMNA gene may even require HTX.48) Patients with LGMD2A, on the other hand, rarely develop CI.49) In isolated case reports, however, CCDs, LVHT, and heart failure have been reported.50) As with LGMD2A, CI in LGMD2B is usually mild51)52) or absent.53) Only rarely may patients present with dCMP.54) In a study of 10 patients with LGMD2C, CCDs and abnormal relaxation pattern in the tricuspid annulus were detected on tissue Doppler imaging.55) Other studies however, did not find CMP in LGMD2C.56) In a study of 32 patients with LGMD2D, two thirds developed CI before the onset of muscular manifestations.57) In a study of 6 patients with LGMD2E, 3 developed fatal dCMP.58) In LGMD2E, systolic dysfunction has been shown to progress during a follow-up of 9 years, whereas ECG parameters may remain unchanged.36) LGMD2I appears to be the LGMD subtype in which CI is most commonly observed. In a study of 23 patients with LGMD2I, almost two thirds had systolic dysfunction, although none had arrhythmias.59) In a study of 10 patients with LGMD2I all had impaired torsion.60) In another study of 7 patients with LGMD2I, 4 had myocardial fibrosis, 1 developed systolic dysfunction, and 1 had diastolic dysfunction.52) cMRI has been shown to be a sensitive method to detect CMP and heart failure in patients with LGMD2I.61) Some patients with LGMD2I may progress to the extent that they require HTX.62) In a study of 32 LGMD2I patients, systolic dysfunction was associated with increased mortality.36) In LGMD2I, systolic function may deteriorate over time while ECG-parameters do not simultaneously worsen. Similar findings were observed in patients with unclassified autosomal recessive-LGMDs.36) CI in LGMD2I may occasionally manifest as acute heart failure.63) dCMP has been reported in single cases with LGMD2M (Table 3).64) In a Dutch study of 24 patients with non-specified sarcoglycanopathy, 17% developed dCMP.65) Patients with LGMD1A, 1C, 1D, and 1E and LGMD2F, LGMD2G, and LGMD2H do not appear to be at risk of developing cardiac disease.66) Mutations in the delta-sarcoglycan gene cause isolated dCMP without skeletal muscle involvement.67)
Facio-scapulo-humeral muscular dystrophy: CI in facio-scapulohumeral musclar dystrophy (FSH-MD) is rare and has been described only in single case reports. These patients developed not only CCDs but also supraventricular and ventricular arrhythmias.68) One FSH-MD patient developed pre-excitation syndrome, supraventricular arrhythmias, and mild myocardial thickening.68) In other patients, short-PR-interval and P-wave abnormalities have been reported.69) In a study of 8 FSH-MD patients, 2 developed elevated P-waves, and three multifocal atrial premature contractions.69) In a single patient with FSH-MD hCMP was reported. In a study of 6 patients with FSH-MD, all subjects had reduced uptake of Tl-201 on Tl-201-SPECT.70) In some FSH-MD patients, sympathetic output may be increased while the parasympathetic output is decreased, as has been demonstrated by heart rate variability analysis.71)
Congenital muscular dystrophies: CMDs are due to mutations in LMNA, LAMA2, COL6A, POMT1, POMGNT1, FKTN, FKRP, LARGE, SEPN1, CHKB, or ITGA7 genes. CI has been reported in various types of CMD. Cardiac involvement, in particular, appears to occur in patients carrying mutations in the LMNA (arrhythmias, dCMP),72) COL6A (dCMP, systolic dysfunction requiring HTX),73)74) POMT1 (aortic root ectasia, systolic dysfunction),75) CHKB (dCMP, systolic dysfunction, congenital heart defects),76) and LAMA2 (dCMP)77)78) genes. Additionally, CI has been reported in patients with Fukuyama congenital muscular dystrophy (dCMP, systolic dysfunction) due to FKTN mutations,79) congenital muscular dystrophy with alpha-dystroglycan deficiency (dCMP, CCDs, mitral regurgitation),80) and in merosin-positive congenital muscular dystrophy (systolic and diastolic dysfunction).81)
Myofibrillar myopathies
Myofibrillar myopathies are caused by mutations in genes which mainly encode Z-disc proteins such as desmin, alpha β-crystalline, myotilin, ZASP, filamin-C, or BAG3.82) Patients with myofibrillar myopathy due to BAG3 mutations may present with CMP at the onset of muscular manifestations.83) In a subset of these patients, cardiac compromise may even precede the onset of muscular manifestations to the extent that HTX is required.83) Some patients carrying BAG3 mutations may present with hCMP with a restrictive filling pattern, QT-prolongation, and, potentially, a rapidly progressive course.84) Rarely, BAG3 myofibrillar myopathy can manifest with dCMP requiring HTX before the onset of muscular manifestations.83) Patients with myofibrillar myopathy and a limb-girdle phenotype due to MYOT mutations may develop CMP only in the late stages of the disease.85) Desmin gene mutations causing generalised weakness and wasting may also be associated with arrhythmias and severe rCMP.86) Patients carrying cypher/ZASP mutations may develop severe dCMP resulting in premature death.87) Patients with non-specified myofibrillar myopathy may present with dCMP and rCMP alongside atrial fibrillation and/or other supraventricular tachyarrhthmyias that respond favourably to recurrent radiofrequency ablation.88) Genetically undetermined myofibrillar myopathy may accompany hCMP, CCDs, and heart failure requiring HTX.89)
Congenital myopathies
Congenital myopathies may be classified according key histological features, such as cores, inclusion bodies, and abnormal protein accumulation, with or without central nuclei, or even based on myofibril size variation. The largest group of congenital myopathies are the core myopathies (central core disease, multiminicore disease), which represent the main non-dystrophic pediatric myopathies.90) Core myopathy due to mutations in the titin (TTN) gene may accompany CMP, atrial septal defects, and LVHT.90) A case report of multiminicore disease-associated LVHT has been described.91) CI has also been reported in multiminicore disease due to MYH7 mutations.92) Rarely, patients with central core disease and intractable dCMP may require HTX.93) A patient with centronuclear myopathy due to a dynamin-2 mutation was also diagnosed with CMP.94) At age 40, this patient subsequently developed left bundle-branch-block (LBBB), three years after which a pacemaker was implanted.94) Early-onset dCMP in centronuclear myopathy may require HTX.95) Infantile myotubular myopathy may accompany the CCDs.96) Congenital fiber-type dysproportion may be associated with CCDs requiring pacemaker implantation.97) Congenital fiber-type dysproportion due to LMNA mutations may also be associated with CCDs.98) CI in nemaline myopathy may manifest as dCMP, hCMP, non-specific CMP, and heart failure.99)100)101) Uncommonly, patients may require HTX, and SCD may also occur on rare occasion.
Distal myopathies
Distal myopathies may be due mutations in MYH7 (Laing), dysferlin (Miyoshi), ZASP (Griggs), VCP, FHL1, TTN (Udd), TOR1AIP, MATR3, or GNE (Nonaka). Only single case reports of patients with genetically confirmed distal myopathy have developed CI. Mutations in the MATR3 gene do not appear to have cardiac manifestations.102) Patients with distal myopathy Welander also do not appear to develop CI. Patients with distal myopathy due to MYH7 mutations may develop dCMP.103)104) An uncommon but important manifestation of CI in distal myopathy due to MYH7 mutations is LVHT.105) X-linked distal myopathy due to FHL1 mutations may accompany hCMP.106) Patients with distal myopathy due to TOR1AIP1 mutations may develop un-specified CMP.107) Distal myopathy due to mutations in desmin may manifest with CCDs requiring pacemaker implantation or, less commonly, with SCD.108) Approximately 10-20% of patients with distal myopathy due to GNE mutations may develop CI.109) Single case reports of patients with non-specified distal myopathy associated histologically with rimmed vacuoles have been reported to present with rCMP, CCDs, or SCD.110)111)112)
Myotonias
Myotonias are clinically characterised by impaired muscle relaxation. Myotonias may be divided into dystrophic and non-dystrophic myotonias. The dystrophic myotonias include myotonic dystrophy type-1 (MD1) and myotonic dystrophy type-2 (MD2).
Non-dystrophic myotonias: Non-dystrophic myotonias include the muscular channelopathies, which only rarely are associated with CI. Isolated cases of patients with hyperkalemic periodic paralysis, however, have been reported in association with CCDs and arrhythmias.113)114) In fact, CCDs appear to be more common in hypokalemic periodic paralysis than in hyperkalemic periodic paralysis.115) In single patients with myotonia congenita Becker, pre-excitation syndrome has been reported.116) CI has not been reported in paramyotonia congenita, potassium-aggravated myotonia, or normokalemic periodic paralysis. In the majority of cases with non-dystrophic myotonias, however, no cardiac abnormalities have been reported.
Dystrophic myotonias: Contrary to non-dystrophic myotonias, the heart is frequently affected in myotonic dystrophies. While both MD1 and MD2 subtypes are affected, cardiac involvement appears to be more common in MD1 with CCDs being among the most common manifestations. The most commonly reported cardiac manifestation is progressive first degree atrio-ventricular (AV)-block, although ventricular arrhythmias do occur and have a significant impact on the overall outcome. AV-conduction disturbances are associated with an increased risk of ventricular arrhythmias.117) In a study of 161 MD1-patients, atrial fibrillation/flutter occurred in 17% and strongly influenced the outcome.118) A high prevalence of ajmalin-induced Brugada ECG-pattern was recently reported in MD1 patients.119) These patients may require a pacemaker or even an implantable cardioverter defibrillator (ICD).120) In MD1-patients, SCD has been commonly reported in the literature.121) In both types of myotonic dystrophy, the myocardium may be affected in the form of dCMP or LVHT.122)123)124) Myocardial involvement may also progress to myocardial fibrosis, thereby manifesting as LGE on cMRI.125) In a study of 129 MD1 patients, CI was found in 55%.126) In this cohort, first degree AV-block was found in 23.6%, second-degree AV-block in 5.6%, right bundle branch block in 5.5%, LBBB in 3.2%, and QTc-prolongation in 7.2%, whereas atrial fibrillation occurred in 4.1%, other supraventricular tachyarrhythmias in 7.3%, and non-sustained ventricular tachycardia in 4.1%.126) In this same cohort, 20% developed systolic dysfunction and 21.7% reduced global longitudinal strain.126) Among patients with congenital myotonic dystrophy, diastolic dysfunction is known to occur.127) More frequently, however, are supraventricular arrhythmias.128) Some patients with congenital MD1 develop AV-block.129) CI in patients with DM2 manifests as conduction disturbances (Brugada-like ECG abnormalities),130) dCMP,131) or LVHT.132)
Metabolic myopathies
Mitochondrial disorders: MIDs are disorders with the highest variability in the incidence of CI. CI in MIDs may not only manifest as CMP, CCDs, or arrhythmias but also as aortic root ectasia,133) coronary artery disease,134)135) pericardial effusion in the absence of heart failure,136) valve abnormalities,137)138) or autonomic dysfunction.139) CI in MIDs most frequently develops after the onset of other systemic manifestations, and only rarely is cardiac disease the initial manifestation of a MID. Among syndromic MIDs, CI most frequently occurs in Kearns-Sayre syndrome, MELAS-syndrome, MERRF-syndrome, chronic progressive external ophthalmoplegia, neuropathy ataxia retinitis pigmentosa, Leigh-syndrome,140)141) or Leber's hereditary optic neuropathy.142)143) CI in patients with primary CoQ-deficiency may manifest as sinus-bradycardia or heart failure.144) CI has been also reported in non-syndromic MIDs.145)146) The most frequent cardiac abnormalities in MIDs are dCMP, hCMP, CCDs, and arrhythmias.147)148)149)150) LVHT is also highly prevalent in MIDs. MIDs, alongside Barth-syndrome, are the disorders with the highest incidence of LVHT. The most frequent manifestations of CI in MIDs, however, remain dCMP, CCDs, and arrhythmias. Despite the multi-organ nature of MIDs, an increasing number of MID patients with intractable dCMP undergo HTX.151)152) The prevalence of SCD in MIDs with CI is unknown, but given the high frequency of involvement of the cardiac conduction system or autonomic fibers is likely quite high. There are also a number of MID patients in whom TTS has been reported.153)
Glycogenoses: CI is common with glycogen storage disease and has been predominantly described in type II, type III, type IV, and type V variants of the disease. Glycogen storage disease type II (Pompe disease) is a rare, autosomal recessive disease in which CI manifests as hCMP, CCDs, supraventricular and ventricular arrhythmias, or heart failure.154) The course of cardiac disease varies considerably between children and adults. Early stages of cardiac involvement responds favourably to enzyme replacement therapy (ERT). CI in glycogen storage disease type-III may manifest as myocardial thickening or hCMP in most patients.155)156) Other cardiac manifestations include myocardial fibrosis, dCMP, or heart failure.157) A single patient with premature coronary artery disease has been described in the literature.158) Some of these patients require HTX.157) Glycogen storage disease IV (Andersen's disease) is due to mutations in the GBE1 gene and may manifest in the heart with dCMP leading to intractable heart failure, which is usually associated with early death during childhood.159) Glycogenosis type V (McArdle) may manifest cardiologically as obstructive hCMP160) and glycogenosis type VII (Tarui's disease) as non-specific CMP.161) Cases of two patients with undetermined vacuolar polysaccharidosis, progressive dCMP necessitating HTX at ages 13 and 14 years have been reported.162)
Lipid storage disease: Disorders of the carnitine cycle or fatty acid oxidation (beta-oxidation) frequently manifest with CI. In a study of 187 patients with beta-oxidation defects, 55% had CI, either manifesting as CMP or arrhythmias.163) Patients with mutations in the HADHB gene may manifest with LVHT and heart failure.164) Patients with very long chain acyl-CoA dehydrogenase deficiency, which manifests with hypoglycaemia, hepatomegaly, myopathy, and rhabdomyolysis, may cardiologically present with CMP or SCD at the onset of the disease.165)166) Patients with neutral lipid storage disease due to PNPLA2 mutations may develop rCMP.167)168) A subset of patients with carnitine-acyl-carnitine translocase deficiency due to a SLC25A20 mutation manifests with non-specified CMP at the onset of the disease.169) Carnitine deficiency may be accompanied by non-specified CMP.170)
Unclassified myopathies: In Barth-syndrome, approximately one-half of patients present with LVHT. Barth-syndrome, together with MIDs, is the disorders with the highest frequency of LVHT. In addition, patients with Barth-syndrome may present with dCMP or heart failure.171) Patients with Barth-syndrome may require ventricular assist devices or HTX. In patients with lysosomal Danon disease, which is due to LAMP2 mutations, hCMP, dCMP, CCDs, arrhythmias requiring prophylactic ICD implantation, and heart failure requiring HTX frequently occur.172) Rarely, these patients may develop LVHT.172) A subset of patients with Danon disease develops SCD.173) CI in Danon disease is usually severe and has strong prognostic implications, and its early diagnosis is, therefore, critical to facilitate timely HTX. In a patient with non-specific muscular dystrophy due to a novel CHKB mutation, dCMP was described.174)
Diagnosis of cardiac involvement in neuromuscular disorders
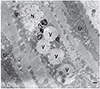
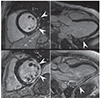
In all NMD patients, CI is important to recognise as it is potentially treatable and may significantly improve outcome with timely, appropriate management. Diagnosing cardiac disease in NMDs is challenging as it may be asymptomatic for a long period of time before becoming clinically evident. Diagnosing cardiac disease in NMDs is particularly difficult if cardiac disease manifests before the onset of neurological manifestations. In cardiologically asymptomatic patients with a clinically evident NMD, CI is usually diagnosed incidentally if one does not proactively search for CI. If patients are cardiologically symptomatic, appropriate diagnostic work-up must to be carried out. The basis of these investigations is a thorough history and careful clinical exam. Instrumental investigations include blood pressure measurement, routine ECG, stress testing, long-term ECG, and echocardiography. Eventually, patients may require coronary angiography, ventriculography, cMRI, stress echocardiography, tissue Doppler imaging, dipyridamole 201Tl SPECT myocardial perfusion imaging, technetium-99m sestamibi (99mTc-MIBI) scintigraphy, or endomyocardial biopsy. Some studies indicate that systolic dysfunction can be more favourably assessed by cMRI as compared to transthoracic echocardiography.175) cMRI detects abnormal distribution and intensity of LGE in 90% of DMD patients and, thus, is a sensitive diagnostic test for identifying early myocardial damage (Fig. 3).175) If the heart is the initial or primrary manifestation of the NMD, endomyocardial biopsy may be helpful to diagnose the underlying condition. Endomyocardial biopsy may show dystrophic changes, reduction or absence of dystrophin in case of a dystrophinopathy, sarcoglycan-deficiency in cases of sarcoglycanopathies, granulofilamentous material in case of a desminopathy, reactive mitochondrial changes in Danon disease, vacuolar degeneration and glycogen deposition in case of a glycogen storage disease, or proliferation and swelling of mitochondria, upregulation of heme oxygenase-1 (an adaptive enzyme to oxidative damage), abnormally shaped mitochondria, or COX-negative fibers in cases of a mitochondrial disorder (Fig. 4). In addition to cardiologic investigations, NMD patients must be evaluated by the pulmonologist and orthopaedic surgeon to rule out pulmonary or orthopaedic causes of CI. Nonetheless, it is important to actively rule out CI in a NMD as soon as the neurological diagnosis is established. Conversely, patients with cardiologic disease of unknown primary should be referred to the neurologist to exclude subclinical or mild NMD.
Treatment of cardiac involvement in neuromuscular disorders
Treatment of CI in NMDs follows the general guidelines for treating specific cardiac disease and generally does not significantly vary from treatment of patients without NMD. Current treatment of CI in NMDs is symptomatic and relies on the avoidance of cardiotoxic drugs, application of cardioactive medications, as well as invasive inteventions, including placement of ventricular assist devices and HTX. If muscle function improves spontaneously or following treatment, it is important to improve overall cardiac function. Improving muscle function without addressing the cardiac aspects of NMDs may aggravate CMP and may, thus, may be harmful.176) Accordingly, the preclinical and clinical attention must assess cardiac function in NMDs.176) Treatment of cardiac disease in NMDs must also consider that patients with advanced involvement of the skeletal muscles can no longer exercise and thus are unable to participate in cardiovascular training and rehabiliation.
Restriction of cardiotoxic drugs
Among NMDs due to a metabolic defect, muscle-toxic cardiac medications, such as beta-blockers, amiodarone, milrinone, or statins should be avoided. In cases of general anesthesia in NMD patients, particularly those with malignant hyperthermia susceptibility or NMDs in which malignant hyperthermia has been previously reported, depolarising muscle relaxants and halogenated volatile anesthetics must be avoided to prevent cardiac arrest or SCD during an episode of malignant hyperthermia.177) For most of the cardiac drugs, however, it is unknown whether they have an adverse effect on nerve or muscle function. With MIDs and beta-oxidation defects, it is uncertain whether cardiac drugs have toxic effects on the mitochondrion. Application of mitochondrion-toxic drugs may not only be beneficial for cardiac disease but may also be harmful to muscle or heart.
Cardiac drugs
In case of heart failure, conservative heart failure therapy with angiotensin-converting enzyme inhibitors, AT-1-blockers, beta-blockers, diuretics, aldosterone-antagonists, must be initiated according to established guidelines. Treatment of CCDs also follows established guidelines. If supraventricular tachy-arrhythmias are also present, digitalis, beta-blockers, or amiodarone may be indicated. If atrial fibrillation occurs alongside the neurologic or muscular disease, digitoxin, beta-blockers, or amiodarone can be given. In cases of brady-arrhythmias, atropine may be a temporary option. In cases of atrial fibrillation or severe systolic dysfunction, oral anticoagulation with vitamin-K-antagonists should be considered. Patients with non-sustained ventricular tachycardia may benefit from beta-blockers. In DMD patients, steroids are beneficial for muscular manifestations but may also be effective in delaying the onset of CMP.178)179) ERT is a beneficial option for hCMP in Pompe's disease.180) A known complication of ERT in Pompe's disease, however, is SCD.180) Infantile or pediatric cases, in particular, may benefit from ERT with a favourable effect on CI.181) Administration of L-carnitine in carnitine deficiency may have a stabilising effect on CMP.170)
Invasive measures
If there is sinus-bradycardia or bradycardious atrial fibrillation in the absence of provocative drugs or if there are pauses >3.5 s, a pacemaker is indicated. If there is coronary artery stenosis, stent implantation and antithrombotic treatment is indicated. In cases of non-sustained ventricular tachycardias unresponsive to beta-blockers, an ICD should be considered. If there is drug-resistant heart failure and a LBBB, a cardiac resynchronisation therapy (CRT)-system should be considered if conventional measures are ineffective. In addition, BMD patients with dCMP, heart failure, sinus tachycardia, and mechanical dyssynchrony benefit from a CRT-system.33) If there are contraindications for an ICD or if the patient refuses ICD implantation, an external defibrillator (LifeVest®) could be an alternative. A LifeVest® can be worn even by pregnant females. Ectopic atrial activity may be treated by radiofrequency ablation.88) If there is severe valve stenosis or insufficiency, valve replacement may be necessary. In a single patient with Fukuyama-type muscular dystrophy, ventriculoplasty because of dCMP had been carried out.182) Myopathies in which implantation of an ICD had been carried out are listed in Table 3. Myopathies in which HTX had been carried out because of intractable heart failure are also listed in Table 3. NMD patients with secondary compromise due to secondary spinal or thoracic deformities frequently benefit from surgical stabilisation of the spinal column.
Preclinical options
Several preclinical therapies are undergoing development, including utrophin up-regulation, stop codon read-through therapy, viral gene therapy, cell-based therapy, or exon skipping.176) Some of these therapies are undergoing clinical trials, but these have predominantly focused on correction of skeletal muscle compromise.176) However, myopathy cannot be treated without simultaneous treatment of CI. Therapies focused on the muscle must also consider and include the myocardium.
Prognosis
In the majority of the cases, the prognosis of cardiac disease in NMDs is fair if the diagnosis is established early, if therapy is adequate, if patients are compliant, and if cardiac abnormalities respond favourably to treatment. However, in the setting of severe ventricular arrhythmias and if the patient is not supplied with an ICD, the ICD is insufficiently shocking, and cardio-pulmonary resuscitation is insufficient, the outcome may be fatal. The prognosis may also be unfavourable in cases of intractable heart failure not amenable to adequate drug treatment, in cases of an unavailable donor heart, or contraindications for a CRT-system or transplantation. In cases of atrial fibrillation, severe heart failure, or embolic events from noncompaction, and contraindications to oral anticoagulation with vitamin-K-antagonists, insufficient anticoagulation or not yet established anticoagulation, thromboembolic events may worsen prognosis and outcome of affected patients.
Discussion
The present review demonstrates that CI predominantly occurs in myopathies and only rarely in neuropathies or transmission disorders. Myopathies most frequently developing CI include the dystrophinopathies, laminopathies, desminopathies, nemaline myopathy, myotonic dystrophies, metabolic myopathies, Danon disease, and Barth-syndrome. It also reveals that CI most frequently includes CMPs (Fig. 5), impulse generation, or CCDs. CI needs to be proactively investigated as it may be subclinical or go unrecognised by the patient. This may be one reason that the prevalence of CI appears low in many myopathies. Another reason may be the overall rarity of the various NMDs. The low prevalence of CI can be also explained by the few systemic cardiac investigations carried out to diagnose CI. This review, in addition, shows that CI in NMDs must be recognised and appropriately treated as early as possible as it significantly determines the outcome of these patients. The earlier that CI in a patient with a NMD is recognised, the better the outcome. An appropriate method to detect early myocardial involvement in NMDs is cMRI. As soon as a NMD is diagnosed, the affected patient must be screened for CI. This is of paramount importance and mandatory irrespective of whether CI is subclinical or symptomatic. The NMDs with a particularly high risk of SCD (Table 3) require close and careful cardiac follow-up so that the therapeutic window is not overlooked.
Survival in NMD patients can be prolonged by oral anticoagulation, heart failure therapy, pacemaker implantation, ICD implantation, or HTX. An increasingly common treatment in drug-resistant heart failure or dCMP is HTX. HTX has the disadvantage of the inevitable use of of steroids and immunosuppressants. Some of these immunosuppressants (e.g. cyclosporine) may secondarily induce myopathy and may, thus, worsen the pre-existing NMD. In addition, neurotoxic drugs, such as chemotherapeutics, may secondarily deteriorate neuropathy. Survival of NMDs with CI is not only dependent on the underlying NMD and the type of cardiac disease, but also on the effectiveness of treatment and avoidance of drugs with a cardio-toxic or myo-toxic effect. Thus, the outcome of patients with NMD and CI is dependent on the medication prescribed, which should be kept to only that which is required. Outcome in patients with LVHT is better if LVHT is isolated compared to LVHT patients with concomitant dCMP or hCMP.183)
In conclusion, CI in NMD is more commonly observed in myopathies, such as the dystrophinopathies, laminopathies, desminopathies, nemaline myopathy, myotonic dystrophies, metabolic myopathies, Danon disease, and Barth-syndrome. Management of CI in NMDs is an evolving field, which has gained increasing scientific attention, has practical implications, and needs to be carefully addressed as its appropriate management can prolong life-expectancy and considerably determines the outcome in most of these patients.



 XML Download
XML Download