

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Despite remarkable advances in cardiac surgery and intervention that have allowed repair of congenital heart defects, pulmonary arterial hypertension (PAH) associated with congenital heart disease remains a major problem. In principle, closure of a large shunt lesions in patients with severe pulmonary hypertension should be performed during early childhood because the patients will progress to an irreversible state with time.
In the past decade, several types of pulmonary vasodilators that can decrease pulmonary arterial pressure have been introduced and have shown promising results.12345) These pulmonary vasodilators appear to be effective in reducing the pulmonary vascular resistance and improving symptoms. Interestingly, this might reverse pulmonary vascular remodelling and enable closure of congenital septal defects in patients previously thought to be in an irreversible or an inoperable state.
We retrospectively analyzed our experience of the stepwise management using targeted medical therapy and surgical/interventional closure in patients with congenital heart disease and severe PAH.
Subjects and Methods
From February 2000 to March 2011, 22 patients with congenital septal defects and severe PAH were managed in Sejong General Hospital. They had borderline pulmonary vascular disease and the pulmonary vascular resistance (PVR) was from 6 to 16 wood units (WU·m2) and/or the pulmonary arterial pressure was more than two-thirds that of the systemic pressure. In cardiac catheterization, pulmonary flow (Qp) was calculated by oxygen consumption6) divided by oxygen content difference between pulmonary vein and pulmonary atery. PVR was calculated by transpulmonar pressure gradient divided by pulmonary flow (Qp). The diagnosis was atrial septal defect (ASD) in 7 patients, ventricular septal defect (VSD) in 6 patients, patent ductus arteriosus (PDA) in 4 patients, atrioventricular septal defect in 4 patients, and VSD type of double outlet right ventricle in 1 patient.
The median age of patients was 16 years (range, 9 months-46 years). Eleven patients (50%) of patients had Down's syndrome. The median follow-up duration from the time of dianosis was 7.4 years (range, 1.4-11.7 years).
Cardiac catheterization was performed in all patients to evaluate the pulmonary hypertension and to determine operability or acceptability of undergoing defect closure.
Based on the biventricular physiology, a patient with a PVR <about 6 WU was considered operable or acceptable for undergoing closure of the defects at our center. In patients with a PVR >6 WU, we usually performed the pulmonary vasoreactivity test with oxygen and patients who showed a decrease in the final PVR to <about 6 WU were considered operable or acceptable for undergoing closure of the defects. The vasoreactivity test was done using oxygen (10 L/min) supplied by mask for 10 min and PVR was calculated in consideration of dissolved oxygen in plasma. Sometimes the balloon occlusion test was performed immediately before complete closure of the defects to confirm the hemodynamic stability after closure and the possible post-procedural outcome. Every decision was taken after an in-depth disscusion with pediatric cardiologists and cardiac surgeons at our center. If it was difficult to arrive at a decision of closing the defect, we avoided prompt closure of the defect and repeated cardiac catheterization.
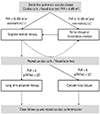
Initial management was selected on an individual basis according to the clinical symptoms and hemodynamic data, such as pulmonary arterial pressure (PAP), PVR, and ratio of pulmnary flow/systemic flow (Qp/Qs). The stepwise approach was performed very slowly and cautiously using targeted medical therapy and surgical/interventional closure (Fig. 1). The targeted medical therapy included phosphodiesterase type-5 inhibitor (sildenafil), endothelin receptor antagonist (bosentan), and/or oral prostanoid (beraprost) as monotherapy or combined therapy.
In this study, we analysed the outcome after dividing the patients into 3 groups according to the initial management. Group 1 included 4 patients (18%) who underwent complete closure of the septal defect. Group 2 included 14 patients (63.6%) who underwent partial closure of the septal defect or patch closure of the septal defect leaving a fenestration. Group 3 included 4 patients (18%) who received medical therapy with pulmonary vasodilators.
This study was approved by the Institutional Review Board of Sejong General Hospital and the requirement for informed consent was waived. Data were expressed as mean±standard deviation or median with ranges for continuous variables.
Results
The diagnoses and the age of patients in each group are presented in Table 1. The patients with a right-to-left shunt or a bidirectional shunt suggesting a more advanced pulmonary vascular disease were more frequently observed in Group 3 compared to the other groups. The patients who had Down's syndrome were more frequently observed in Group 1 and Group 2 than in Group 3.
PVR in patients of Group 1 was relatively lower when measured in room air than that in other groups, and decreased markedly to less than 6 WU on the vasoreactivity test (Fig. 2A and B). However, Group 2 and Group 3 showed higher PVR and a partial or no response to pulmonary vasodilator. The amount of a left-to-right shunt (Qp/Qs) was higher in Group 1 compared to Group 2 and Group 3 (Fig. 2C).
Among the patients in Group 1 who underwent complete closure of septal defects, 3 patients underwent surgical closure of ventricular septal defect (VSD), atrioventricular septal defect (AVSD), and PDA; and 1 patient underwent percutaneous closure of PDA. There was no mortality and the New York Heart Association (NYHA) Functional Class was improved in all of the patients. After defect closure, the mean ratio of peak right ventricular pressure/peak aortic pressure {p(RV/Ao)} or ratio of peak pulmonary arterial pressure/aortic pressure {p(PA/Ao)} was <0.3 (range, 0.32-0.35). The echocardiographic findings did not show any evidence of PAH or right ventricular enlargement during the mean follow-up period of 7.4 years (range, 1.4-11.7 years). In two Group 1 patients, follow-up cardiac catheterization showed a normal PVR (<3 WU) and PAP at 4.2 years later after complete closure.
In Group 2, 14 patients underwent partial closure of septal defects or patch closure of orginal defects leaving a fenestration that functioned as a pop-off valve. Fenestration creation of atrial septum was performed in 5 patients with VSD or PDA. Partial ASD closure was performed in 9 patients, partial VSD closure was performed in 2 patients, and partial closure of PDA by banding was performed in 1 patient. There was one case of early mortality that developed bacterial pneumonia and severe pulmonary hypertension after surgery and the patient eventually died of septic shock. The other patients showed a postoperative p(PA/Ao) or p(RV/Ao) of <0.5. Still persistent PAH after surgical management necessitated subsequent medical therapy in 5 patients who underwent follow-up cardiac catheterization at 4.45 years later after partial closure in Group 2 (Fig. 3).
Group 3 included one case of complete AVSD, 2 cases of PDA, and one case of VSD. Because these patients had unfavorable hemodynamic data and poor vasoreactivity, and seemed to be in an inoperable clinical state, they initially received targeted medical therapy without cardiac surgery (Table 2). In this group, the patients showed a low Qp/Qs and a high PAH that did not change with administration of pulmonary vasodilation. They also showed mild resting desaturation, consistent with right-to-left shunting on echocardiography before the initiation of targeted medical therapy. Generally, we performed follow-up cardiac catheterization annually or biennially. The mean duration from targeted medical therapy to closure of defects was 3.6 years (range, 15-96 months). Several years later, the direction of the shunt changed to a purely left-to-right shunt on echocardiography and the PVR decreased and Qp/Qs increased on serial cardiac catheterizations. These patients underwent successful closure of defects and subsequent long-term targeted medical therapy with the expectation of a further decrease in the PVR. All patients showed a decrease in PVR after targeted medical therapy (Fig. 4A) compared to baseline data. As well, PVR after administration of pulmonary vasodilators for vasoreactivity test was decreased markedly up to the acceptable level to perform the septal defect closure (Fig. 4B). In addition, the mean Qp/Qs increased from 1.6 to 3.6 (Fig. 4C), and chest X ray showed increase in the cardiomegaly. Of these patients, 3 underwent surgical partial closure of defects (PDA and VSD) or ASD fenestration, and 1 underwent pulmonary atery banding. The p(PA /Ao) or p(RV/Ao) was 0.3 in the PDA patient, 0.5 in the VSD patient, and 0.7 in the PA banding patient. Patients who underwent cardiac surgery showed improvement in the NYHA functional class.
Two years later, 2 patients underwent percutaneous closure using the Amplatzer device (Mendelssohn Avenue Golden Valley, MN, USA) after confirmation of safety with balloon test occlusion of the residual fenestration. Fig. 5 shows the serial changes in PVR over time after the combined therapy including pulmonary vasodilator medications, cardiac surgery and subsequent percutaneous closure of residual defect in these 2 patients who were previously considered to be in an irreversible state of pulmonary vascular disease. PVR was markedly decreased after targeted medical therapy with pulmonary vasodilators and did not increase again, even after subsequent surgical and percutaneous closure. One patient plans to undergo follow-up cardiac catheterization, and another patient is being treated with pulmonary vasodilators and is awaiting to undergo percutaneous complete septal defect closure in the future.
There was one case of early mortality (4.5%), and there was one case of late mortality (4.7%), which was related to aggravation of systemic lupus erythematosus. All patients showed improvement in the NYHA functional class (FC); FC I was seen in 90% of patients (n=18) and FC 2 was seen in 10% of patients (n=2). Eventually, complete closure of septal defects was achieved in 36.8% of patients (n=7), and partial closure of septal defects or closure of septal defects leaving a fenestration was achieved in 63.2% of patients (n=12). During the management, 50% of patients (n=11) were given pulmonary vasodilators as a preoperative conditioning therapy and/or as postoperative long-term therapy for residual pulmonary hypertension. Twenty percent of patients (n=4) of patients required continuous medication with pulmonary vasodilators because of residual pulmonary hypertension.
Discussion
A recent major breakthrough in the treatment of patients with PAH has been the availability of oral endothelin antagonists and the application of sildenafil, a type-5 phosphodiesterase inhibitor.7)
The decision to perform closure of congenital septal defects largely depends on the amount of the shunt and the status of pulmonary circulation. The early repair of large septal defects in patients with severe pulmonary hypertension is crucial toprevent irreversible pulmonary vascular changes. Patients diagnosed with septal defects later in life and thoase with pulmonary vascular disease are at a high risk of sustained pulmonary hypertension, right heart failure, and hypertensive crisis immediately after surgery.8)9) Right-to-left shunting in the presence of severe pulmonary hypertension sustains the cardiac output at the expense of arterial hypoxemia, and is a mechanism for decreasing the right heart pressure in the presence of right ventricular failure. Generally, when right-to-left shunting with cyanosis is present, one has to seriously question the indications and safety of septal defect closure.8)
In the past, operability or acceptability to undergo septal defect closure was often decided based on pathological findings in lung biopsy.10) Nowadays, the decision is based more on the severity of pulmonary hypertension and the degree of vasoreactivity of the pulmonary circulation with the use of pulmonary vasodilators.11) An acute reduction in the mean pulmonary arterial pressure >10 mmHg with a resultant mean pulmonary arterial pressure of ≤40 mmHg without a fall in the cardiac output is considered a positive vasoreactive response in adult.12) Howerver, positive vasoreactivity does not indicate operability and evaluation of this criteria has not been studied adequately in children with congenital heart disease. In the present study, the possible operable state was defined as a baseline PVR <about 6 WU or a decrease in the final PVR after the administration of pulmonary vasodilators to <about 6 WU.13)
There have been significant advances in the development of therapies for PAH. Before 1996, treatment options for PAH were limited to conventional therapies including digoxin, diuretics, anticoagulation, supplemental oxygen whenever indicated, calcium channel blockers in selected patients, and lung or heart-lung transplantation. Intravenous epoprostenol was then included in the treatment algorithm for PAH as a bridge to transplantation. Over the past decade, prostacyclin analogues, selective and nonselective endothelin receptor antagonists, and phosphodiesterase type 5 inhibitors have been introduced. Potent oral pulmonary vasodilators that allow long-term administration have become available with promising results.1) These targeted PAH medical therapies might have the potential to reduce the pulmonary vascular resistance, lower the perioperative risk and possibly widen the range of cases amenable to repair.14) In this study, 4 patients who were initially considered as inoperable underwent successful closure of defects after achieving acceptable PVR following the targeted medical therapy.
Irreversible pulmonary vascular obstructive disease increases the perioperative risk, lowers the chances of benefiting from the procedure, and increases the risk of postoperative right ventricular failure. Nevertheless, despite the presence of long-standing pulmonary vascular disease with evidence of significant vascular remodelling/obstruction, Eisenmenger patients often respond favorably to advanced therapy.14) Also, it has been suggested that certain pulmonary vasodilators have antiproliferative effects that cause reverse remodeling in the pulmonary circulation.15) In this study, the response to chronic vasodilator therapy suggests that PAH-targeted therapy may play the role of a conditioning therapy in carefully selected patients with a possible late septal defect closure.
There are serious concerns about the long-term results and the tolerability of septal defect closure after targeted medical therapy remains unknown. There have been some case reports showing promising results, although the follow-up duration was relatively short.16171819) However, the follow-up duration was relatively short. Once the defect is closed and the PVR is still high or it increases again, a pathophysiologic situation that is similar to idiopathic pulmonary hypertension could develop, which is associated with a much worse long-term outcome compared to that in Eisenmenger patients. Although the early postoperative risk can be overcome, right ventricular failure may develop many years later.
Appropriate selection of patients is very difficult because the response to targeted medical therapy is not uniform. During the early period of this study, we were very reluctant to decide upon surgical/interventional closure and frequently waited and reevaluated the patients. However, recently we adopted a more aggressive approach including use of a pulmonary artery band and the administration of targeted therapy at the same time in patients with a large VSD.20)
Partial closure of a septal defect or creation of atrial fenestration was performed to allow decompression of the right ventricle during periods of raised PVR,21)22) during both the early postoperative period and long-term follow-up. In adult cases, we made a 6-8 mm residual shunt for partial closure or fenestration. In pediatric cases, the size of residual shunt was decided on by the surgeon.
Subsequent complete closure of septal defect with a percutaneous approach can be achieved if PVR decrease and is persistently low in response to long-term targeted medical therapy in the patients.
This study has inherent limitations. This is a retrospective study comprising a small and heterogeneous group of patients. The duration of observations was relatively short. The results of cardiac catheterization that we regarded as decisition criteria for management may have sources of error in flow and resistance calculations, such as sampling errors, measurement errors, assumptions of oxygen consumption and pulmonary venous saturation, and approximations of mixed venous samples. Furthermore, even though patients have the same results of cardiac catherization, responses for intervensions that included pulmonary vasodilators, surgical operations, and percutaneous procedures would be different. Therefore, making a general guideline for patients with congenital heart disease and pulmonary vascular disease is difficult.
In conclusion, targeted medical therapy may be effective in reducing pulmonary vascular resistance in patients with near systemic pulmonary arterial pressure who were previously thought to have irreversible pulmonary vascular disease. Also, a stepwise approach with combined targeted medical therapy and surgical/interventional closure, the appropriate use of fenestration, and performing the balloon occlusion test before complete occlusion may help to achieve more improved outcomes in patients who have congenital heart disease and borderline pulmonary vascular disease. We suggest a management algorithm for patients with congenital heart disease and borderline pulmonary vascular disease in Fig. 6.
Judicious decision-making and close follow-up of patients are necessary because the long-term efficacy of targeted medical therapy for PAH is not yet known, and tolerability of long-term administration of this therapy has not been established.23) The diagnostic and therapeutic management for patients with congenital heart disease and borderline pulmonary vascular disease should be careful and tailored to the individuals. Also, long-term and continuous monitoring for them is required.






 XML Download
XML Download