

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Diabetes mellitus (DM) is one of the major chronic diseases affecting about 300 million people worldwide, and the number of affected people is increasing. It is estimated that there will be about 450 million people with diabetes by 2030.1) Cardiovascular diseases represent a major cause of morbidity and mortality in patients with diabetes. Particularly, coronary artery disease (CAD) is the major etiological factor for death in diabetic patients.1) Although CAD and ischemia are the major causes of heart failure in diabetic patients, the distinct entity 'diabetic cardiomyopathy (DCM)', was first described by Rubler et al.2) who reported autopsy data from four diabetic patients with congestive heart failure and normal coronary arteries in 1972.
Today, the occurrence of DCM is still a misunderstood "entity". The definition of DCM has been initially confined to the presence of abnormal myocardial performance or abnormal structure in the absence of epicardial CAD, hypertension and significant valvular disease. Other facets of this entity possibly consist of cardiomyopathy because of diabetic macroangiopathy and/or microangiopathy, myopathy, cardiac and/or autonomic neuropathy, sequelae of antidiabetic agents and arrhythmias, and diabetes itself.3)4)Studies have shown that heart failure is increased about 2-3 times in diabetic patients independent from the etiology. The prognosis of heart failure is much worse in diabetic patients.5) However, in the absence of hypertension and CAD, the mechanism of heart failure in diabetic patients is not fully understood. To date several mechanisms have been implicated in the pathogenesis of DCM. This paper will attempt to summarize 41 years of knowledge on this topic.
Cardiac Structural and Functional Changes
Left ventricular hypertrophy
Diabetic cardiomyopathy is characterized by ventricular dysfunction in the absence of hypertension and ishemic CAD. Data from the Framingham Heart Study indicated that an increased left ventricular (LV) mass and wall thickness were more prominent symptoms in diabetic female patients.6) However, the Strong Heart Study showed that both men and women with diabetes had higher LV masses and wall thickness.7) Studies with magnetic resonance imaging (MRI) showed that insulin resistance and impaired glucose levels were consistently and independently associated with LV hypertrophy and LV mass to LV end diastolic volume ratio.8)
Hypertrophy of cardiomyocytes appears to be a frequently observed feature but not a prerequisite of DCM. It is considered as an affect of insulin which induces hypertrophy and cellular growth. Data from animal studies9)10) have also revealed that myocardial hypertrophy was most likely to be related with insulin resistance and high insulin levels. This is a typical characteristic of type 2 DM, whereas myocardial fibrosis was likely to be related to early hyperglycemia, which is a typical characteristic of type 1 DM.9)10) Increased aortic pulse pressure is related with LV hypertrophy, and it has been shown that impaired glucose tolerance is associated with increase aortic pressure in patients without CAD and hypertension.11)
Diastolic dysfunction
Diastolic dysfunction is the basic hemodynamic characteristic and the earliest findings of DCM can be detected by using imaging techniques. The prevalence of this abnormality may vary according to the used imaging technique, diagnostic criteria, and definitions among several clinical studies. LV diastolic dysfunction can be basically assessed by using the criteria in the consensus statement on the diagnosis of heart failure with normal LV ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology.12) Also, improved glycemic control in DM demonstrated that the LV diastolic dysfunction might be reversible during the early stages. Furthermore, LV diastolic dysfunction in DCM may also progress to a systolic dysfunction and could result in reduced LV ejection fraction (EF) in years. Therefore, it is important to detect LV diastolic dysfunction in diabetic patients, both for early diagnosis and treatment of DCM, and prevention of the occurrence of systolic dysfunction. According to our clinical experience, dyspnea is the first symptom of patients with diabetes and diastolic dysfunction. Diastolic dysfunction with preserved LV EF also presents with symptoms of systolic dysfunction. However, routine investigation into the etiology of dyspnea in preserved EF usually do not end up with DCM. These patients may be easily misdiagnosed due to this unsuspecting clinical entity.
LV diastolic dysfunction by the use of flow and tissue Doppler techniques was observed in 45-75% of asymptomatic, and normotensive patients with type 2 DM.13)14) Previous studies reported that diastolic dysfunction was observed both in type 1 and type 2 diabetic patients.15)16) Tissue Doppler imaging (TDI) is more sensitive to show diastolic dysfunction than conventional echocardiography. TDI enables measurement of the movements of mitral valve at specified myocardial locations near the mitral annulus. It was shown that mitral annulus early (E) filling was significantly lower in normotensive diabetic patients than in the control group.17) Similarly diastolic dysfunction was observed both by invivo echocardiography and in exvivo tissue samples from animal models of type 2 DM, such as ob/ob and db/db mice and Zucker diabetic fatty rat.18)
Systolic dysfunction
Systolic dysfunction is characterized by the disability of LV to pump a sufficient amount of blood to the body. In early stages of DCM, LV EF can be normal and it is defined as a heart failure with preserved LV EF. Currently, shortened LV ejection time, decreased peak systolic velocity (S'), and smaller LV fractional shortening can be the detectable parameters of systolic dysfunction. In some studies, patients with DM had smaller LV fractional shortening than those subjects with normal glucose tolerance.9)19) Low-quality echo-Doppler devices and conventional 2D echocardiographic techniques may miss or are incapable of detecting subtle systolic dysfunction. This is due to the fact that classical 2D echocardiography can determine only LV circumferential functions, and can not estimate LV longitudinal functioning. However, TDI and strain rate imaging techniques are able to estimate LV function in longitudional, radial and circumferential ways. They are able to detect systolic dysfunction more accurately in diabetic patients. However, patients with diabetes and low LV EF values should be evaluated for CAD first. In the case of normal coronary arteries with hypertrophied, fibrotic and weak contracted LV could help to diagnose DCM.
In early stages of DCM, LV dysfunction can be induced by exercise in asymptomatic patients. In some studies it has been reported that during exercise peak systolic velocity, peak exercise stroke volume index, and cardiac index were significantly lower in both uncomplicated Type 1 DM and type 2 DM patients than in the control group. In these studies two groups had normal LV echocardiographic functions at rest.9)20) Also in animal models with ob/ob mice, it was shown that contractile reserve was reduced by inotropic stimulation.21)
Pathological Mechanisms
Impaired calcium handling
Calcium (Ca) is a key ion in excitation-contraction coupling of cardiomyocytes. Impaired calcium handling in the cardiomyocytes is thought to be the mechanistic hallmark of DCM. Calcium handling is regulated by several transporters inside the cell. Ca enters the cell through L-type Ca channels, and induces calcium release from sarcoplasmic reticulum through the ryanodine receptors, which is the main mechanism for contraction. For relaxation to occur, calcium ions eject from the cell through Sarcoplasmic reticulum Ca pump (SERCA), increasing Na/Ca exchanger and sarcolemmal Ca ATPase.
Prolongation of action potential duration due to decreased Ca efflux from cytosol is consistently observed in diabetic cardiomyocytes. Calcium homeostasis is altered by reduced activity of SERCA, Na/Ca exchanger and Ca ATPase. Studies conducted in both type 1 and type 2 diabetic rats showed reduced activity of ryanodine receptors, and depressed Ca efflux from cytosol.22) Decreased SERCA activity resulting in impaired relaxation and increased action potential duration, should correlate with clinical findings of diastolic dysfunction.23) SERCA activity can be decreased by either reduction in protein levels or by post translational modification due to non-enzymatic glycosylation. Decreased sensitivity of contractile proteins to calcium may also contribute to the contractile dysfunction of cardiomyocytes, and was shown in animal and human studies.24)25) However, a recent study by Falcao et al. reported increased Ca sensitivity in diabetic rats.26)
Altered metabolism
Free fatty acids and glucose are major metabolic energy sources of the heart. The healthy heart is able to switch rapidly between these sources, and accommodate to the different physiologic and pathologic conditions. In the fasting state, the heart utilizes free fatty acids, whereas in the postprandial state or under stress and ischemia, glucose becomes the preferred substrate for energy production. However, in a diabetic heart, utilized substrate for metabolism shifts to higher level of free fatty acids and decreased level of glucose because of insulin resistance and hyperlipidemia.
Glucose uptake is decreased by down-regulated expression of glucose uptake transporter-4 in response to insulin resistance. This in turn leads to reduced rates of glycolysis and glucose oxidation. Reduced glucose oxidation and hyperlipidemia causes increased fatty acid oxidation. Fatty acid oxidation is augmented not only by the elevation of free fatty acids but also by activation of peroxysomal proliferator activated receptor alfa (PPARα). Free fatty acids activate PPARα and this activates several enzymes to increase beta oxidation and increases expression of pyruvate dehydrogenase (PDH) kinase 4. This further suppresses glucose utilization by decrasing PDH. Fatty acid oxidation also requires high levels of oxygen compared to glucose metabolism, and causes relative cardiac ischemia. During ischemia aerobic glycolysis shifts to anaerobic glycolysis which results in an accumulation of lactate and acid metabolites. The toxic intermediates resulting from fatty acid oxidation can impair Ca homeostatis and myocyte contraction; this phenomenon is called lipotoxicity. Increased free fatty acid uptake further causes lipid accumulation in cardiomyocytes. Rijzewijk et al.27) also showed that myocardial triglyceride content assessed by magnetic resonance spectroscopy has been increased in uncomplicated type 2 DM, and was associated with impaired LV diastolic function, independent of age, body mass index, and visceral fat. Lipid accumulation also leads to accumulation of ceramide which is associated with contractile dysfunction and apoptosis of cardiomyocytes.
A recent study28) investigated the ability of melatonin in reducing metabolic risk factors and cardiac apoptosis, induced by diabetes. Streptozotocin was injected into male rats, after diabetic induction, melatonin (10 mg/kg i.g.) was administered orally for 21 days. Melatonin effectively ameliorated diabetic myocardium injury, apoptosis, reduced the metabolic risk factors, and modulated important steps in both extrinsic and intrinsic pathways of apoptosis.
Increased oxidative stress
Reactive oxygen species (ROS) formation from mitochondria and cytosol is another major contributor to the progression of DCM. ROS are known to cause oxidative stress through oxidation of critical biomolecules including proteins and nucleic acids {ribonucleic acid and deoxyribonucleic acid (DNA)}. Lipids lead to the damage and dysregulation of the cellular structure, physiological and metabolic machinery, ultimately cause pathophysiological alterations in the cells, tissues, organs, and the entire organism. ROS can cause protein and lipid damage by oxidation and can cause an increase in DNA damage due to reduced activity of DNA repairing pathways. Excessive ROS production has been implicated in cardiac hypertrophy, fibrosis, contractile dysfunction and heart failure.29) Moreover, interactions between ROS and nitric oxide generates nitrotyrosine species. It has also been shown that increased 3-nitrotyrosine was associated with increased apoptosis in both animal and human cardiomyocytes.30)31) A study conducted in rats provided strong evidence that Nox4 is an important source of ROS in the left ventricle, and that Nox4-derived ROS contribute to cardiomyopathy at early stages of type 1 diabetes.32) ROS scavenging systems have been shown to be efficacious in reducing diabetes induced cardiac dysfunction. Increased level of metallothionein, catalase and manganese superoxide dismutase, reduce deterioration of myocardial dysfunction and improve diastolic function in animal models.33)
Activation of renin angiotensin system (RAS) is related to increased activity of nicotinamide adenine dinucleotide phosphate, which causes elevated ROS formation, and myocardial fibrosis and cell death. One study demonstrated that inhibition of nuclear factor kappa b signaling in the heart prevents diabetes induced cardiac dysfunction through preserved calcium handling and inhibition of the cardiac renin-angiotensin system in rats.34) It has been shown that, angiotensin II stimulates various vascular growth factors that leads to ROS production.35) Blockade of RAS activation ameliorates interstitial fibrosis and improves myocyte contraction.36)
Remodeling of extracellular matrix
Hyperglycemia seems to be a central element in the pathogenesis of DCM. Prolonged exposure of proteins to glucose over a long time causes them to undergo a series of non-enzymatic reactions, eventually forming advanced glycosylation end products (AGEs). The AGEs are a stable form of cross-linked collagen and may be responsible for the stiffness of arterial walls and myocardium, endothelial dysfunction and atherosclerotic plaque formation. Positive correlations of serum level of AGEs with ventricular isovolumetric relaxation time, arterial stiffness and carotid intimal thickness have been shown in diabetic patients.37)38)39) A recent study conducted in transplant hearts by Jerzy Nozynski et al.40) reported that chronic heart failure increases AGE deposition mostly in veins, whereas type 2 DM predisposes arterioles to AGE accumulation. Receptors for advanced glycosylation end product (RAGE) also participate in the pathogenesis of DCM. It was shown that LV myocardial contractility were improved in RAGE-blocked diabetic mice, indicating that RAGE is involved in mechanisms related to the contractile properties of the myocytes, which in turn is probably related to decreased function of SERCA.41)
Strategies to prevent AGE formation, block activation of AGE receptors, or break the AGE-protein cross-links, have been suggested as potential therapies to attenuate pathogenic influences of AGEs on myocardial function in diabetes. Treatment with an aminoguanidine, an inhibitor of AGE formation, prevents progression of ventricular dysfunction in diabetic rats.42)
Endothelial dysfunction
Vascular endothelial cells play a pivotal role in the maintenance of cardiovascular homeostasis. Normal and healthy endothelium produce various vasodilators, such as nitric oxide, prostacyclin, bradykinin, and endothelium-derived hyperpolarizing factor, and also produce vasoconstrictors such as endothelin and angiotensin II. Exposure to high levels of glucose under diabetic conditions can damage the physiological properties of endothelium and alter its physiological processes. This can cause enhanced permeability, leukocyte adhesion, and reduced fibrinolysis.43)
Conditions of insulin resistance caused by type 2 diabetes, atherosclerosis, and endothelial dysfunctions are all known to induce the expression of the pro-inflammatory cytokine, tumor necrosis factor (TNF)-α, which can increase the expression of vascular and intercellular cell adhesion molecules, and promote adherence of monocytes.44) TNF also reduces endothelial nitric oxide synthase expression and interferes with nitric oxide production. Moreover, under hyperglycemic conditions, the coronary circulation gets exposed to increasing amounts of acetylcholine, which paradoxically constricts the coronary arteries, thereby leading to coronary vasospasm.
Mitochondrial dysfunction
Mitochondria of the heart play crucial roles in several important physiological and pathophysiological functions such as, production of energy, maintenance of intracellular Ca levels, regulation of apoptosis, mitoptosis, formation of ROS, modulation of cellular oxidative stress, thermoregulation, autophagy and modulation of cellular signaling events.
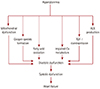
The critical role of cardiac mitochondrial dysfunction in the pathogenesis of cardiac diseases, including ischemia reperfusion damage and diabetic myocardial defects, has been highlighted. Although several crucial factors, such as alterations in lipid metabolism, insulin resistance, and altered adipokine secretion have been recognized to play significant roles in diabetic cardiomyopathy (LV dysfunction), evidence is mounting for the role of cardiac mitochondrial abnormalities/dysfunction in the pathogenesis of diabetic cardiomyopathy.45) Alterations or imbalances in the cardiac mitochondrial bioenergetics (energy metabolism) has shown to be critical factors in the pathogenesis of diabetic cardiomyopathy.46) Hyperglycemia during diabetes has been recognized as a key factor in the diabetes-induced myocardial defects, including diabetic cardiomyopathy through structural abnormalities in the myocardium (cardiac hypertrophy, fibrosis, myofibril defects, and cardiomyocyte aberrations) and myocardial mitochondrial defects, such as mitochondrial swelling and fewer number of mitochondria. Oxidative stress has also been associated with the diabetes-induced myocardial structural alterations (Fig. 1).
Diagnosis
The DCM is a diagnosis of suspicion. Unfortunately, there is no widely accepted method for the diagnosis of DCM. The best approach is detection of myocardial dysfunction, and exclusion of other heart diseases, which may cause myocardial structural and functional changes. Clinically, it may take several years for heart failure to develop in diabetic patients. So, it is very essential to demonstrate the abnormality before symptoms of heart failure begin.
Echocardiography is a available, reliable and noninvasive imaging tool to demonstrate early fuctional changes of LV. Early diastolic and late systolic dysfunction can be shown by echocardiography. Normal echocardiographic findings at rest do not exclude the diagnosis of DCM. LV dysfunction detectable by TDI, during exercise or stress, may also be the earliest sign of DCM.
Other diagnostic methods such as computed tomography (CT), single photon emission CT, and MRI can be used for detection of myocardial dysfunction. Assesment of interstitial fibrosis and steatosis by using delayed gadolinium enhancement cardiac MRI is possible but its diagnostic value has not been established.
Prevention and Treatment
Hyperglycemia is considered as a main factor for development of DCM. Hyperglycemia results in AGEs production, cardiac collagen accumulation and oxidative stress. Prevelance of cardiac dysfunction differs in several studies. A study by Kiencke et al.47) conducted in diabetic adults, without any evidence of structural heart disease, revealed that DCM was present in 48% of the patients who had an increased risk for functional deterioration, and possibly cardiovascular events, during follow-up.47) To date, it is unclear why certain diabetic patients develop cardiomyopathy. The possible explanation may be the severity of hyperglycemia and duration of DM, which well correlates with the presence of DCM. The amount of accumulated AGEs in the myocardium was found to be related with the duration of diabetes. However, results of studies investigating the association of glycemic control with cardiac functions are controversial. Study by von Bibra et al.48) reported that diabetic patients show significant improvement in diastolic velocities with strict glycemic control. However, meta analyses including United Kingdom Prospective Diabetes Study, (the Prospective Pioglitazone Clinical Trial in Macrovascular Events, Action to Control Cardiovascular Risk in Diabetes, Intensive Blood Glucose Control and Vascular Outcomes in Patients with Type 2 Diabetesand Veterans Affairs Diabetes Trial trials showed no significant differences when there was a combined analysis for a non-fatal stroke and cardiovascular and all-cause mortality, between the intensive glucose lowering strategy and the standart treatment strategy.49)
The benefits of the inhibiton neurohormonal activity with angiotensin converting enzyme inhibitors, angiotensin receptor blockers and beta-blockers have been evidenced in DCM. Hence, contribution of the angiotensin receptor 1 to fibrosis was shown in diabetic models; inhibition of receptor activity improves LV function. A meta analysis included 28 trials, beta-blockers with carvedilol, metoprolol, and bisoprolol which reduced all-cause mortality and sudden death in heart failure patients. Metoprolol was prominent in the reduction of sudden death, compared with carvedilol or bisoprolol.50)
Conclusion
Diabetes mellitus itself can cause cardiac dysfunction without the formation of atherosclerosis and hypertension. Clinical and experimental studies with diabetes have demonstrated that DCM was associated with cardiac structural and functional changes. Early diastolic and late systolic dysfunction correlate with glycemic status and duration of diabetes. Cardiac dysfunction in asymptomatic diabetic patients can be detectable by various techniques. It is imperative to make an early diagnosis and reduce disease progression. Therapeutic agents toward the specific metabolic and structural derangements of DCM are encouraging, but there is still no specific treatment strategy to manage DCM. Further clinical research focused on the mechanisms of DCM will clarify the therapeutic approach to the prevention and treatment of this entity.
 XML Download
XML Download