

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The sinoatrial node (SAN) automaticity is essential for maintaining normal cardiac function. Sick sinus syndrome is an abnormality involving the generation of the action potential by the SAN and is characterized by an atrial rate inappropriate for physiological requirements. The sick sinus syndrome occurs in 1 of every 600 cardiac patients older than 65 years and accounts for approximately half of implantations of pacemakers in the United States.1) A better understanding of the mechanisms of SAN automaticity and sick sinus syndrome is therefore clinically important. The SAN automaticity is maintained by synergistic actions of a "voltage clock" mediated by voltage-sensitive membrane ionic currents such as the hyperpolarization-activated pacemaker current (If)2) and a "Ca2+ clock" mediated by rhythmic spontaneous sarcoplasmic reticulum Ca2+ release.3) While extensive work has been performed to document the interactions of voltage and Ca2+ clocks of the SAN in animal models, translating these findings to human patient care is difficult. Our review will focus on the translational studies of human SAN function.
Pacemaker Hierarchy and the Importance of Ca2+ Clock in an Intact Sinoatrial Node
Cardiac automaticity at the organ level is very complex. In addition to cellular mechanisms, integrative anatomical and physiological factors are involved in cardiac pacemaking. The intact SAN is a heterogeneous structure that includes multiple different cell types interacting with each other.4)5)6) The Ca2+ clock in the superior SAN is primarily responsible for rate acceleration during sympathetic stimulation.7)8)9)10)11) However, the relative importance of the voltage and Ca2+ clocks for pacemaking in different regions of the SAN, and in response to neurohumeral stimuli such as β-agonists, may be different. Indeed, activation maps in intact canine right atrium (RA) have shown that the SAN impulse origin is multicentric,12 and sympathetic stimulation predictably results in a cranial (superior) shift of the pacemaking site in human and dogs.13)14) Based on evidence from isolated SAN myocytes, late diastolic Cai elevation relative to the action potential upstroke is a key signature of pacemaking by the Ca2+ clock.15)16)17)18)19)20) The same phenomenon could provide insights into the relative importance of the Ca2+ and voltage clock mechanisms in pacemaking in intact RA tissue,7) subsidiary pacemakers21)22) or diseased state.23)24)
Mapping of Earliest Atrial Activation Site in Humans
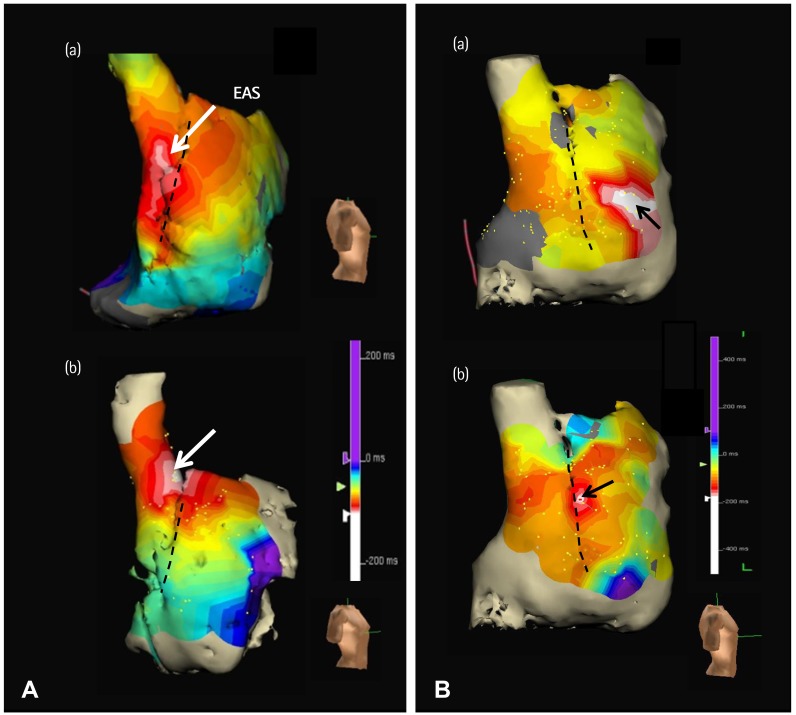
Schuessler et al.14) reported in a canine model that sympathetic stimulation in general tends to induce a cranial shift in the location of the pacemaker within the pacemaker complex. With the development of 3 dimensional (3D) endocardial electroanatomical mapping techniques, it is possible to define the activation patterns within the human atria to accurately locate the earliest atrial activation site (EAS).25) If the findings in canine models are applicable to humans, then the superior SAN should vigorously respond to sympathetic stimulation and serve as the dominant pacemaker during sympathetic stimulation. Indeed, EAS of control human subjects shifted cranially during isoproterenol infusion (Fig. 1A).26) These findings indicate that in both canine models and humans, the superior SAN is primarily responsible for heart rate acceleration during sympathetic stimulation in patients with normal SAN function.
Sinoatrial Node Function in Patients with Atrial Fibrillation and Symptomatic Bradycardia
Atrial fibrillation (AF) is associated with significant electrophysiological and structural remodeling of the atria, and is often associated with sick sinus syndrome.27)28) The SAN dysfunction may be reversible after successful catheter ablation of AF.29) In dogs, persistent (>2 weeks) rapid atrial pacing and chronic AF resulted in SAN dysfunction, as evidenced by prolongation of the SAN recovery time and decreases in the intrinsic heart rates.30) Unresponsiveness of the Ca2+ clock in the superior SAN to sympathetic stimulation is a characteristic finding in dogs with AF and heart failure.31)32) Consistent with that found in a canine model,31) the patient with AF with SAN dysfunction had impaired heart rate acceleration and absence of upward shift of the EAS during isoproterenol stimulation (Fig. 1B). These findings suggest that Ca2+ clock malfunction underlies these abnormal physiological responses to isoproterenol infusion.
The Cranial Shift of the Earliest Atrial Activation Site and Sinoatrial Node-Right Atrium Propagation
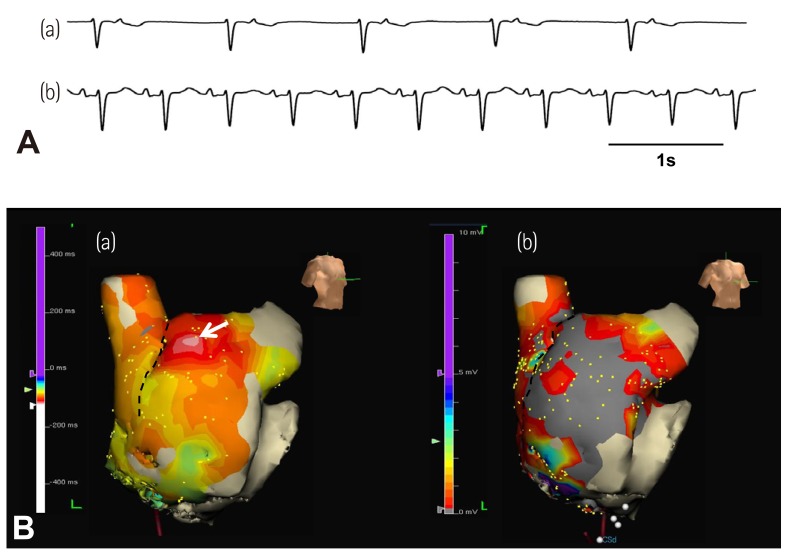
Previous studies have shown that the impulse from the SAN propagates into RA through an upper and a lower exit site.33) It is possible that isoproterenol preferentially shifts the exit site to the upper portion of the SAN instead of the activation of the superior SAN by the activation of Ca2+ clock. Because the extracellular electrograms cannot be used to differentiate SAN activation and RA activation, the shifting of SAN exit sites and the shifting of the actual pacemaking sites may look the same on the 3D map. The data from our human 3D mapping study cannot be used to differentiate these two mechanisms.26) However, we found that isoproterenol infusion reactivates the pacemaker in the superior SAN in several patients with extensive RA fibrosis and junctional rhythm at baseline (Fig. 2). In these patients, changing the sinoatrial node-right atrium exit pathway is clearly not a mechanism of upward shift of the EAS. Moreover, in other patients with upward shifting of EAS during isoproterenol infusion, the conduction between SAN to RA occurred at multiple directions, without evidence of conduction delay along the crista terminalis or the septum. Based on these findings and the mapping of the Ca2+ clock activity in the canine models, we propose that the pacemaker hierarchy is likely to be created by differential responses of Ca2+ clock to sympathetic tone at different portions of the SAN rather than changing the exit from a fixed pacemaking site.
Sinoatrial Node Dysfunction and Amiodarone
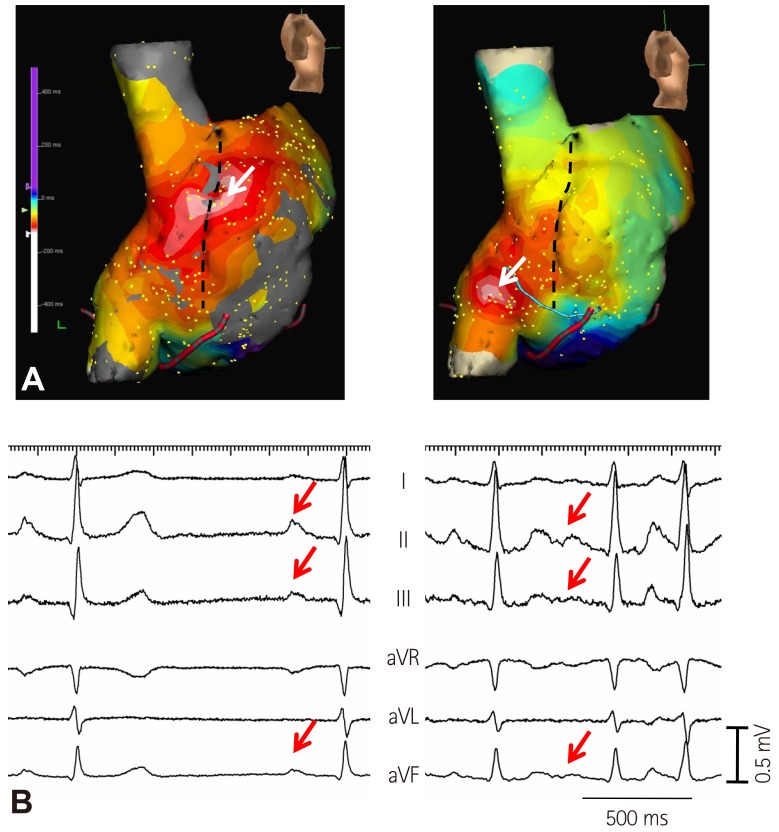
As the elderly population continues to expand, AF is becoming an increasingly common medical condition.34)35)36)37) Amiodarone is the most frequently used agent for maintaining sinus rhythm in patients with AF. Amiodarone impairs SAN function in one-third of patients and is associated with an increased risk of pacemaker insertion.38) Our study also showed that amiodarone induced SAN dysfunction in one-fourth of patients without SAN dysfunction at baseline.39) Amiodarone caused impaired heart rate acceleration and impaired cranial shift of EAS after sympathetic stimulation (Fig. 3).39) Therefore, Ca2+ clock malfunction of the superior SAN might be related with impaired heart rate acceleration and cranial shift of the pacemaking site. Amiodarone inhibits multiple ion channels (Ito, IKr, IKs, ICa, INa) and is a beta adrenergic blocker.40) Because amiodarone is a potent blocker of Ica,40) it may suppress the Ca2+ clock in the SAN.41) In addition, amiodarone can inhibit the sympathetic activity. A downward shift of the EAS has been reported in human patients after esmolol infusion.25) Therefore, the beta blocking effects of amiodarone may also influence the location of the EAS. Amiodarone may induce hypothyroidism, and this effect may, in part, decrease SAN function.38) Finally, Turker et al.42) recently showed that amiodarone inhibits the small-conductance calcium-activated K (SK) channel. Because the SK channel is important in SAN and atrioventricular node function,43)44) SK channel inhibition may affect SAN automaticity.
Unicentric versus Multicentric Activation
Sanders et al.45) previously reported that multicentric activation is found both in normal control and heart failure. However, the EASs are more numerous in normal control (average 4 sites) than in heart failure (average 2.5 sites). They also reported that the SAN complex in patients with SAN dysfunction is more often unicentric, localized to the lower crista terminalis at the site of the largest residual voltage amplitude.45) We also found that most healthy control patients had multicentric SAN activation pattern (Fig. 1A), and that the number of EAS in AF patients is smaller than that in healthy controls (Fig. 1B). A reduction of the EAS suggests that there are fewer backup pacemaking sites to respond to sympathetic stimulation, which could be a sign of SAN dysfunction.
P-Wave Morphology and the Earliest Atrial Activation Site
The P-wave morphology has been used to identify the origins of focal atrial tachycardias with a high sensitivity and specificity.46)47)
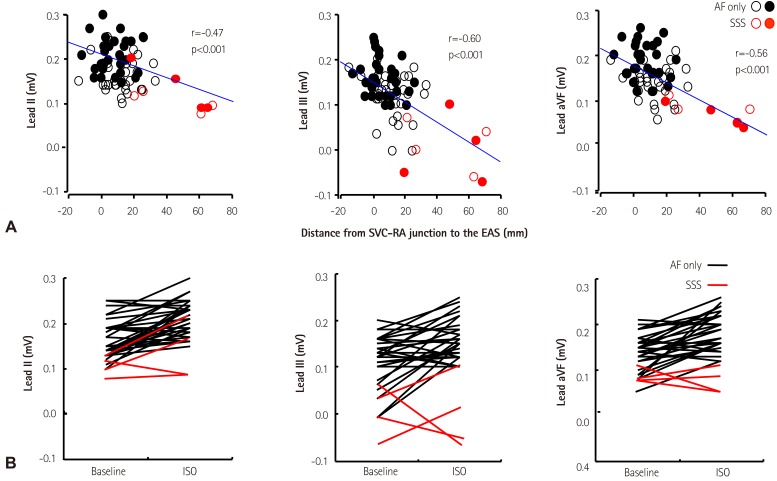
Fig. 4 shows that compared with sinus rhythm (arrows), atrial tachycardia originating from the inferior part of crista terminalis had smaller amplitude of P-wave in leads II, III, and aVF (broken arrows). We found that the distances from the superior vena cava-right atrial junction to the EAS were negatively correlated with the amplitude of P-wave in inferior leads. If the EAS was shifted to the superior part of crista terminalis or superior vena cava, the P-wave amplitudes were increased in most of the patients without SAN dysfunction.39) In contrast, the shift of the EAS to the inferior part of crista terminalis or inferior vena cava was related with decreased P-wave amplitude in inferior leads in patients with SAN dysfunction (Fig. 5A). These findings suggest that the change of P-wave morphology in inferior leads during isoproterenol infusion can be used to assess the superior SAN responsiveness to isoproterenol infusion. Patients with SAN dysfunction showed low amplitude of P-waves at baseline and during sympathetic stimulation (Fig. 5B).
Comparative Efficacy of Testing for Symptomatic Bradycardia
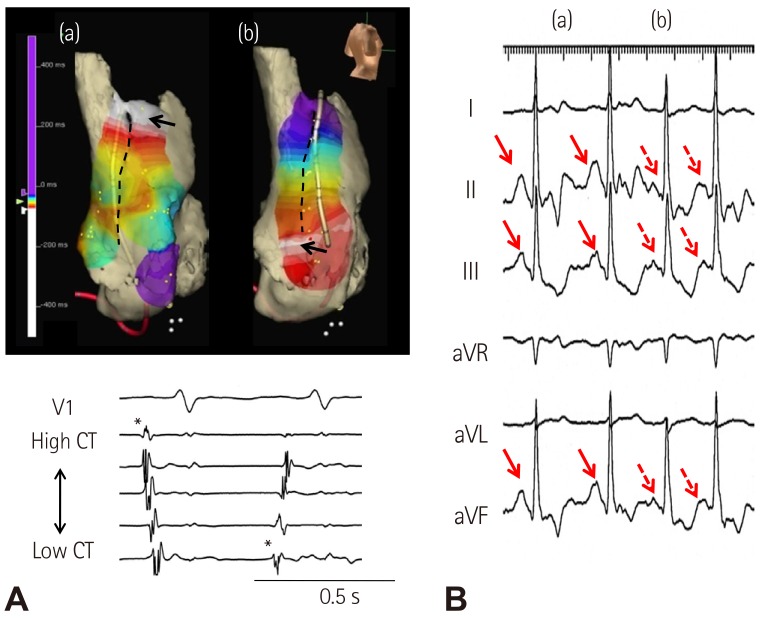
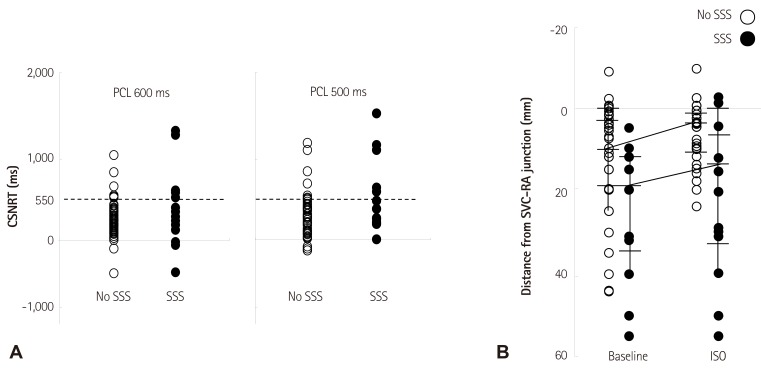
We analyzed the comparative efficacy of corrected sinoatrial node recovery time (CSNRT) and 3D mapping in differentiating AF patients with and without sinus bradycardia. CSNRT>550 ms is considered a positive CSNRT test. The failure of superior SAN to serve as EAS during isoproterenol infusion is considered a positive 3D mapping test (Fig. 6). The sensitivity, specificity, positive predicative efficacy, and negative predicative efficacy of the CSNRT test are 35%, 89%, 45% and 84%, respectively (Fig. 6A), and of the 3D mapping test are 78%, 78%, 47% and 93%, respectively (Fig. 6B). The 3D mapping test was twice as sensitive but slightly less specific than the CSNRT test in detecting AF patients with sinus bradycardia. However, both tests have limitations as they are invasive and time consuming tests.
Recently, we analyzed the poor increase in inferior P-wave amplitude during sympathetic stimulation for the diagnosis of sick sinus syndrome. The poor increases of P-wave amplitude of lead aVF (<0.1 mV) during isoproterenol infusion showed a sensitivity of 78% and specificity of 89% for the diagnosis of sick sinus syndrome. Finally, the combined algorithm using CSNRT of >550 ms and poor increases of P-wave amplitude of lead aVF showed further improved diagnostic accuracy (sensitivity of 89% and specificity of 76%).
Recent Basic Researches on Sinoatrial Node Automaticity
Calmodulin-dependent protein kinase II (CaMKII) has emerged as a central regulator of physiological SAN responses and a key determinant of SAN dysfunction.48) The calcium and CaMKII is especially important for physiological "fight or flight" SAN beating rate responses.49) Inhibition of CaMKII in SAN does not affect baseline heart rate, but reduces heart rate increases in response to physiological stress. Excessive CaMKII activity, as occurs under pathological conditions such as heart failure, ischemia, and diabetes, can promote intracellular Ca2+ overload and reactive oxygen species production. Nicotinamide adenine dinucleotide phosphate oxidase is activated in conditions with increased angiotensin II, leading to oxidation of two methionine residues of CaMKII, rendering the enzyme autonomously active. Increased CaMKII activation leads to SAN cell death, reducing the threshold volume of automatic cells of the SAN and increasing non-excitable tissue in the form of fibrosis. CaMKII-induced apoptosis results in SAN cell loss, which disrupts the normal source-sink balance leading to sinoatrial node dysfunction.50)51) In a canine heart failure model, increased expression of adenosine receptors within the SAN region together with increased fibrosis have been reported to be related with the loss of synchrony and sinoatrial node dysfunction.52) A source-sink mismatch caused by cell loss has been observed in a mouse model of diabetes and in ankyrin-B syndrome.53)54)
Conclusion
The voltage and Ca2+ clocks jointly regulate SAN automaticity. In various models of sick sinus syndrome, the dysfunction of superior SAN during sympathetic stimulation was consistently observed. In human 3D mapping, superior SAN was identified as the leading pacemaker site during sympathetic stimulation. However, unresponsiveness of superior SAN to sympathetic stimulation was commonly observed in patients with sinus dysfunction. 3D mapping, which showed unresponsiveness of the superior SAN or inferior P-wave amplitude, was more sensitive than classic CSNRT in identifying patients with sinus dysfunction. These novel tools obtained from basic research might help to diagnose sick sinus syndrome
Sources of Funding
This study was supported in part by research grants from the Basic Science Research Program through the National Research Foundation of Korea funded by the Korean Heart Rhythm Society (2011-3), the Ministry of Education, Science and Technology (NRF-2010-0021993, NRF-2012R1A2A2A02045367), a grant of the Korean Healthcare technology R&D project funded by Ministry of Health & Welfare (HI12C1552), United States National Institutions of Health grants R01 HL71140 and P01 HL78931, the Indiana University Health/Indiana University School of Medicine Strategic Research Initiative and a Medtronic-Zipes Endowment of the Indiana University.
 XML Download
XML Download