

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Cardiomyopathy is a serious disease associated with high mortality and morbidity. The cause of cardiomyopathy in children is currently poorly understood. Only one third of children know the cause of their disease at the time of diagnosis, while the remaining two thirds are classified as idiopathic.1) Recent advances in biochemical and molecular genetic techniques have contributed to improving our understanding of the metabolic disorders causing cardiomyopathy. Inborn errors of metabolism account for only 5% of all pediatric cardiomyopathy, and 15% of those are due to known causes, but they are of particular interest to clinicians because many have disease-specific treatments.2)
Identification of underlying causes of cardiomyopathy with subsequent disease-specific treatments may lead to improved outcomes. Primary carnitine deficiency (PCD) is one of the treatable causes of cardiomyopathy since complete resolution of the heart disease is possible if treated early.3-5)
In this review article, an overview of the clinical characteristics, diagnosis, treatment, and long-term outcome of PCD is provided with an emphasis on cardiac involvement.
Carnitine Metabolism and Fatty Acid Oxidation
Carnitine (beta-hydroxy-gamma-trimethylaminobutyric acid) is a naturally occurring hydrophilic amino acid derivative produced endogenously in the kidneys and liver or derived from meat and dairy products in the diet. In normal human omnivores (non-vegetarians), approximately 75% of carnitine sources come from the diet while the rest is provided by endogenous synthesis.6) Carnitine is eliminated via urine as free carnitine and acylcarnitines, but more than 95% of carnitine filtered in the kidney is reabsorbed by the proximal tubules therefore maintaining the homeostatic balance of carnitine in the body.7)
Long-chain fatty acids are important energy substrates for the myocardium and other muscle tissues. However, they cannot freely diffuse through the mitochondrial inner membrane. Carnitine plays an essential role in the transfer of long-chain fatty acids into the mitochondria for subsequent β-oxidation (Fig. 1). Deficiency of carnitine blocks the mitochondrial oxidation of fatty acids to carbon dioxide in all tissues and to ketones in the liver, and leads to lipid accumulation in the cytosol. Since skeletal and particularly cardiac muscles depend on fatty acid oxidation for most of their energy, these tissues can be expected to be the most severely affected by carnitine deficiency.4)
Clinical Carnitine Deficiency Syndrome
Two distinct carnitine deficiency states have been reported, which include "primary" and "secondary" carnitine deficiency.8)
Primary carnitine deficiency (OMIM 212140), also known as carnitine uptake defect, carnitine transporter deficiency or systemic carnitine deficiency, is an autosomal recessive disorder of the carnitine cycle that results in defective fatty acid oxidation. The incidence of PCD is about 1 : 40000 with an approximate carrier rate of 1%, although this frequency has been reported to be higher in the Faroe Islands, an isolated archipelago in the North Atlantic.9-11) The disease is caused by heterogeneous mutations of SLC22A5 which encodes the high-affinity carnitine transporter OCTN2 in the plasma membrane.12)13) OCTN2 is strongly expressed in the myocardium, skeletal muscle, renal tubules, fibroblasts, intestine and placental tissue.14)15) The Na+-coupled active transport of carnitine via OCTN2 is essential for the maintenance of high carnitine concentrations inside cells. With the help of plasma membrane carnitine transporters, intracellular carnitine concentrations are maintained 20 to 50 times greater than those of the extracellular space. SLC22A5 mutations can affect carnitine transport by impairing the maturation of transporters to the plasma membrane. Defects of the OCNT2 transporter protein result in urinary carnitine wasting, low serum carnitine levels, and decreased intracellular carnitine accumulation.14-16) As a consequence, impaired oxidation of long chain fatty acids leads to deficient energy generation as well as reduced ketogenesis, particularly during fasting or stress, and accumulation of long-chain fatty acids in the cytoplasm of affected tissues. The presentation of affected patients can vary widely with respect to the age of onset and organ involvement, from asymptomatic to lethal cardiac manifestations. The clinical symptoms usually consist of episodes of hypoketotic hypoglycemia, hepatomegaly, muscle weakness and cardiomyopathy with congestive heart failure.
Secondary carnitine deficiencies may be hereditary or acquired. Secondary carnitine deficiency can be caused by a number of organic acidemias, defects of fatty acid oxidation and of the carnitine cycle. However, PCD is the only genetic defect in which carnitine deficiency is the cause, rather than the consequence.17) Carnitine deficiency can also be acquired as a result of liver disease, renal disease (Fanconi syndrome, renal tubular acidosis), premature birth, dietary insufficiency (chronic TPN, malabsorption, soybean based infant formula), and drug therapy such as valproate acid and pivampicillin.18)
Cardiac Involvement of Carnitine Defficiency
In 1980, the first case of PCD was described by Chapoy et al.19) in a 3-year-old boy. The initial presentations were hypoketotic hypoglycemic encephalopathy, hepatomegaly, and cardiomegaly when he was 3 months old. This boy responded dramatically to oral L-carnitine supplementation, as evidenced by increased muscle strength, relief of cardiomyopathy, and complete repletion in the liver. Since then, more and more evidence has demonstrated that PCD is a treatable cause of cardiomyopathy in children.
There is a considerably broad phenotypic range associated with PCD, ranging from early infantile decompensation to adults who are predominantly asymptomatic. Of the various phenotypes manifested by this condition, the most common presentation of the disease is progressive cardiomyopathy with or without skeletal muscle weakness beginning at 1-4 year of age. A comprehensive review of 61 literature-reported PCD cases revealed an exclusive cardiac manifestation frequency of 62.3% with a very low likelihood of simultaneous occurrence of cardiac and metabolic manifestations.20)
Cardiomyopathy is the most common clinical manifestation in children with PCD, which include dilated cardiomyopathy and hypertrophic cardiomyopathy. More specifically, Tripp et al.3) reported a severe form of familial dilated cardiomyopathy similar to endocardial fibroelastosis due to systemic camitine deficiency. Histologic evaluation at autopsy revealed massive lipid accumulation with endocardial fibrosis in both ventricles in their patient. The incidence of dilated cardiomyopathy was higher than hypertrophic cardiomyopathy in children with PCD.21) In our previous study, however, we observed a mild degree of ventricular hypertrophy in some patients presented with dilated cardiomyopathy.22) Stanley et al.23) reviewed the presenting features of 20 infants and children with defects in carnitine uptake. The most commonly noted clinical feature at initial presentation was progressive cardiomyopathy, which occurred in 12 of the 20 patients in the cohort. The average age of cardiomyopathy presentation is 2-4 years of age, indicating that it takes a long period of time for the manifestations of severe carnitine deficiency in the heart and skeletal muscle to appear. Affected individuals are generally normal at birth and may be asymptomatic for several years until they develop overt signs of cardiomyopathy and congestive heart failure. Their cardiac function does not respond to inotropes or diuretics. If the condition is not correctly diagnosed and no carnitine is supplemented, progressive heart failure eventually leads to death.
Cardiomyopathy, which is frequent in infantile patients, is not commonly seen in adults. The prevalence of PCD in the Faroe Islands is the highest reported in the world (1 : 300).10)11) From a nationwide screening program in the Islands, 76 adult patients identified with PCD were predominantly asymptomatic with normal cardiac structure and function.24) However, the reported number of untreated adults with PCD is quite low elsewhere. Among previously reported cases, dilated cardiomyopathy was present only in one adult patient and excluded in most of the others.25)26) Moreover, SLC22A5 mutations are no more frequent in adult patients with cardiomyopathy than in the general population.27)
Cardiac arrhythmia, although rare, is another clinical manifestation in patients with PCD. In a literature review of 42 adult patients, cardiac arrhythmias were present in 5 of them (12%). Prolonged QT interval with syncope was described previously in two young women with PCD, but short QT intervals and ventricular fibrillation has also been reported in PCD patients.26)28) Ventricular fibrillation without overt cardiomyopathy may be a presenting symptom of PCD. A case report described a 15-year-old girl without overt cardiomyopathy presented with cardiac arrest due to ventricular fibrillation, suggesting that carnitine uptake deficiency should be considered as a potential cause of unexplained cardiac arrhythmias.29) Bradycardia and atrial arrhythmias are also possible in PCD patients. The underlying mechanisms of arrhythmia in PCD have not yet been established.
A significant cohort of patients with PCD do not present symptoms in infancy or early childhood and remain asymptomatic into their adulthood. These observations are derived from the experience of expanded newborn screening programs that identified maternal PCD in mothers who were for the most part minimally symptomatic or asymptomatic.30) Unfortunately, the first clinical manifestation in asymptomatic individuals may be sudden death. Several sudden and unexpected deaths have been reported in PCD patients.31) Severe ventricular arrhythmias are suspected as the cause of unexpected death in patients harboring the defect.
Carnitine deficiency can be exacerbated by other causes, such as pivalic acid exposure. Treatment with antibiotics containing pivalic acid results in increased excretion of carnitine in urine and reduced carnitine concentration in serum. A previous report showed a strong association between pivalic acid exposure and severe complications including encephalopathy and lethal cardiac arrhythmia in patients with PCD.32)
Although there are few special echocardiographic features in patients of PCD with cardiomyopathy, it has been reported that some of them present with characteristic changes on the electrocardiogram (ECG). Tripp et al.3) first described these changes in 1981 in a school aged girl of PCD with cardiomyopathy and cardiac syncope. He noted a bizarre T-wave enlargement (60 to 80 mm) similar to that seen in extensive posterior-wall ischemia as well as hyperkalemia in the mid-precordial leads. Since then, Waber et al.4) and Matsuishi et al.33) respectively reported similar electrocardiographic findings in children with PCD and cardiomyopathy. It is even more interesting that Waber et al. observed peaked T waves reverting to normal after L-carnitine treatment. Thus, he pointed out that tall, peaked T waves in the mid-precordial leads should raise the question of systemic carnitine deficiency. More recently, Tein et al.34) reported four children with PCD and cardiomyopathy, three of whom had strikingly peaked T waves on the ECG which also disappeared with L-carnitine therapy. In our previously reported six cases of PCD and cardiomyopathy, similar T wave alterations were found in five of them and disappeared after L-carnitine therapy. Moreover, we found that all our six patients presented with short QT interval owned to the significantly shortened or even disappeared ST-segment, and the intervals normalized after L-carnitine therapy (Fig. 2).22) Retrospective data analysis of the publications mentioned above also demonstrated tall peaked T waves and short QT intervals simultaneously. Labarthe et al.28) pointed out that these ECG changes are associated to ventricular tachycardia and sudden death of PCD patients, but the underling mechanisms are still unknown. A recent study showed that long-chain acylcarnitines could lead to action potential repolarization abnormalities by regulating the rapidly activated delayed rectifier potassium channel (Ikr/HERG) in vitro.35) This might be a possible explanation why PCD patients present with highly elevated T waves and shortened QT intervals similar to those seen in patients with hyperkalemia. It is still not clear whether the presence of short QT intervals is specific in patients with PCD compared to the other cardiomyopathies. Further studies need to prove the correlation between short QT intervals and sudden cardiac death in this population.
Cardiac Manifestation in Heterozygotes
Heterozygous carriers usually have about 50% carnitine transport activity in fibroblasts and can have borderline low plasma carnitine levels due to increased urinary loss.16) This raises a question of whether they may be susceptible to cardiac dysfunction, left ventricular hypertrophy and arrhythmias in the context of aging and additive cardiac risk factors. Although, in general, patients who are heterozygous for SLC22A5 mutations have been thought to be asymptomatic, Garavaglia et al.36) described a 4-month-old male heterozygous infant with poor growth, vomiting, and left ventricular hypertrophy as shown by ECG and ECHO at 8 months of age. Studies of animal models have shown that such heterozygotes have a higher incidence of cardiomyopathy with aging and may be at risk for cardiac abnormalities when additional cardiac burdens are imposed.37)38) A genetic epidemiological study in Japan showed that the prevalence of heterozygotes for SLC22A5 mutations was 1.01%, and the echocardiography analysis revealed that they were predisposed to late onset benign cardiac hypertrophy (odds ratio 15.1, 95% CI 1.39-164) compared with wild-types.9) However, a recent study examined the prevalence of heterozygotes for SLC22A5 mutations in 324 patients with cardiomyopathy and demonstrated that heterozygosity for PCD is not more frequent in patients with unselected types of cardiomyopathy and is unlikely to be an important cause of cardiomyopathy.27) Finally, heterozygosity for PCD mutations has been associated with cardiac arrhythmias that resolve after carnitine treatment.39) In any case, considering that about 1% of the population is heterozygous for this potentially treatable condition, more studies with careful follow-ups are deserved in the future.
Genotype-Phenotype Relationship
No association between genotype and phenotype in PCD was found in previous studies.40) Patients with identical mutations can have different ages of onset and different types of clinical presentations.41) Even siblings with the same mutation have different ages of onset and different progressions of disease pointing to the presence of clinical heterogeneity.21) However, a recent study suggested that nonsense and frameshift mutations are typically associated with lower carnitine transport and are more prevalent in symptomatic individuals whereas missense mutations and inframe deletions, which are more prevalent in asymptomatic individuals, may result in proteins with retained residual carnitine transport activity.42) Another study suggests that the R245X and V295X mutations of SLC22A5 may be associated with cardiomyopathy as the only clinical phenotype.43)
Diagnosis
The clinical criteria for PCD include severe reduction of plasma or tissue carnitine levels (free carnitine <5 mM, controls 25-50 mM); evidence that low carnitine levels impair fatty acid oxidation; correction of the disorder when carnitine levels are restored; and the absence of other primary defects in fatty acid oxidation. Diagnosis can be further confirmed by demonstrating reduced carnitine transport in skin fibroblasts in the patient or by mutation analyses of the SLC22A5 gene. The introduction of tandem mass spectrometry (MS/MS) for the analysis of plasma acylcarnitine profile has greatly facilitated the identification of patients with defects in fatty acid β-oxidation and has unquestionably been the most striking advance in newborn screening. With the advent of expanded newborn screening, more patients with milder or asymptomatic presentations are getting diagnosed with PCD. Therefore, the actual prevalence of this condition in the general population may be much higher than originally anticipated.44)
Primary carnitine deficiency should be differentiated from other causes of carnitine deficiency. Severe secondary carnitine depletion is also a characteristic feature of many inborn errors of metabolism, some of which may present as cardiomyopathy. These include a number of organic acidemias, defects of fatty acid oxidation and of the carnitine cycle. In all these disorders, analysis of urine organic acids, plasma amino acids and acylcarnitine profile, in conjunction with the clinical presentation, allows a definitive diagnosis.45)
The identification of abnormal cardiac muscle function should help clinicians to consider all possible causes, including genetic and acquired disorders of energy metabolism. It has been proposed that all children with idiopathic cardiomyopathy should be evaluated for carnitine deficiency, as treatment with high-dose carnitine supplementation can reverse the pathology. In our previous study, carnitine deficiency was detected using tandem mass spectrometry in six of seventy-five children with dilated cardiomyopathy; PCD was further confirmed by mutation analyses of the SLC22A5 gene.22)
Some adults dying from ventricular fibrillation may have suffered from undiagnosed PCD. Although rare, PCD should be routinely investigated as a cause of cardiac arrhythmias since arrhythmia episodes can be prevented by oral carnitine supplementation. PCD should be suspected in patients with cardiomyopathy and electrocardiographic tall peaked T waves in the mid-precordial leads, with or without short QT intervals.
Therapy and Prognosis
Treatment of this disorder with L-carnitine is highly effective in correcting cardiomyopathy and muscle weakness as well as any impairment in fasting ketogenesis. The Food and Drug Administration first approved L-carnitine in 1985 for the treatment of PCD. Primary treatment involves supplementation of oral L-carnitine. Typically a high dose, 100-400 mg/kg/day, that is divided in three doses is required. The dose of carnitine should be adapted to each individual patient by serial measurements of plasma carnitine levels. Treatment should include, in addition to L-carnitine, supportive and other therapies as indicated by the condition of the patient. No side-effects are noted for L-carnitine supplementation except occasional diarrhea or fishy body odour. This is usually self-limiting and resolved by reducing carnitine dosage.45)
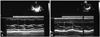
Treatment with oral L-carnitine at pharmacologic levels is quite effective in treating cardiomyopathy and muscle weakness in these children. Within a few weeks of therapy, there was a dramatic resolution of severe congestive heart failure and myopathy in all patients; within a few months of therapy, there was a reduction of heart size toward normal. On the other hand, suspension of treatment leads to the recurrence of signs and symptoms. In our previous study, PCD was identified in 6 patients presented with progressive cardiomyopathy, characterized by enlargement of the left ventricle and greatly reduced left ventricular ejection fraction (LVEF). After 6-month treatment of L-carnitine, LVEF returned to normal in all 6 patients. Left ventricular end-diastolic diameter decreased in all of the 6 patients and returned to normal levels in 3 patients (Fig. 3). No clinical signs or symptoms were present in all 6 patients. The only complications of L-carnitine therapy was intermittent diarrhea in 1 patient.22)
The low number of reported adult PCD patients in the literature has made it difficult to assess the risks and susceptibility to complications in patients suffering from PCD. It is not clear whether the apparently asymptomatic cases of the defect require the same degree of therapy as this is a recent observation and inavailability of sufficient follow-up data.
Currently, there is no consensus on the treatment of SLC22A5 heterozygotes with L-carnitine supplementation. Careful prospective follow-up of SLC22A5 heterozygotes for carnitine deficiency with more sensitive modalities of cardiac testing is needed to assess whether long-term carnitine deficiency increases the risk of cardiac dysfunction.39)
Carnitine levels may be nonspecifically depressed in patients with cardiomyopathy, regardless of the underlying cause. This constitutes the basis for administration of L-carnitine to all children with dilated cardiomyopathy. Such a strategy has been shown to be useful in a multicenter retrospective study involving North American Centers and in a small prospective uncontrolled study from India.46)47) Moreover, several studies have examined the efficacy and safety of L-carnitine treatment for cardiomyopathy in adult patients without PCD, which suggested that L-carnitine therapy may improve the functional status of patients with moderate to severe heart failure attributable to dilated cardiomyopathy.48) However, the role of carnitine depletion in cardiomyopathy and heart failure in patients without PCD, as well as the potential of L-carnitine supplementation as an element in the treatment of these disorders, is still under investigation and debate.
The prognosis for individuals with PCD depends on the age, presentation, and severity of symptoms at the time of diagnosis.44) Early detection of these disorders before the occurrence of irreversible organ damage may lead to more effective treatment and more favorable outcomes.
Until now, very few cases of cardiomyopathy due to PCD with long-term follow-up have been reported in the literature. Several children have survived into their third decade and appear to be doing well, even though L-carnitine treatment raises muscle carnitine concentrations to only 5-10% of normal.49)50) Two patients have died suddenly of cardiac arrest in their second and third decades when L-carnitine supplementation was discontinued, suggesting that continued treatment is essential.50)51)
Up to now, there have been no formal surveillance guidelines for patients with PCD. Lifelong treatment with L-carnitine and avoidance of fasting or prolonged exercise are required. Regular echocardiogram, ECG and plasma carnitine level evaluation is mandatory during follow-up visits.


 XML Download
XML Download