

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Ageing is associated with the increasing stiffness of the large arteries. Studies have shown that increased arterial stiffness, which is a clinical parameter that reflects the degree of arterial ageing, is a significant predictor of cardiovascular disease independent from blood pressure and other cardiovascular risk factors. Arterial stiffness is mainly associated with a patient's age and is also significantly influenced by other conditions that induce oxidative stress. Recent studies revealed the molecular mechanism as well as pathologic findings of the arterial ageing processes. This article will cover the pathophysiology of arterial ageing, recent evidence regarding the treatment of arterial aging and novel treatment targets that are investigated.
Ageing and Vascular Changes
Structural changes
Arteriosclerosis is the pathological term that is used frequently to describe arterial ageing. However, arteriosclerosis is a broad definition encompassing atherosclerosis, Mönckeberg medial calcific sclerosis, and arteriolosclerosis.1-3) However, none of these terms can describe exactly all of the pathological changes of the ageing arteries, for example, age related medial degeneration and sclerosis.4) Compared to healthy young vessel, aged vessels are characterized by increased reactive oxygen species and vascular inflammation that results in endothelial dysfunction. Subsequently, vascular smooth muscle cells (VSMC) migrate into the subendothelial space then proliferate and cause an accumulation of collagen and proteoglycan in the intima.5) On the other hand, the number of VSMCs in the media layer decreases due to apoptosis. Moreover, elastin is degraded in the media and the space is replaced by collagen accumulation and calcification. The elastin degradation of the media layer is one of the major pathological findings of arterial ageing and is a major reason for the increased stiffening of the large arteries with ageing. Various pathways including inflammatory cytokines, adhesion molecules, matrix metalloproteinases (MMPs) and transforming growth factor-β are involved in the ageing process of the major arteries.6)7)
Functional changes
The major functional change that occurs with arterial ageing is the loss of distensibility. Functional and structural mechanisms account for the decreased distensibility and increased stiffness. The functional deterioration of the aged arteries are characterized by endothelial dysfunction8)9) with significant decrease in the acetylcholine induced nitric oxide dependent vasodilation.10) Also, the number of receptors in the vessel and the affinity decreases and consequent decrement of β2-receptor dependent vasodilation occurs.11) As the large arteries stiffen the velocity of the pulse wave increases. The measurement of this velocity, called the pulse wave velocity (PWV), is a common surrogate marker for assessing the degree of arterial ageing. The loss of large arterial distensibility is associated with increased forward amplitude, which will increase the systolic pressure. Also, increasing PWV will result in the early return of the reflected wave and increased augmented pressure. This will subsequently lead to increased systolic blood pressure and pulse pressure which lead to more stiffening and thickening of arterial walls in a vicious cycle.11) Even though increased arterial stiffness is a major mechanism for the pathogenesis of systolic hypertension in the elderly, arterial stiffness itself is an independent risk factor for the major cardiac events independent from blood pressure.12) Studies have demonstrated a significant increase in cardiovascular events in patients with carotid femoral PWV of 12 meter/sec or higher.13) As such, the European Society of Hypertension classifies a person with carotid-femoral PWV higher than 12 meter/sec as a high risk hypertensive patient.14)15)
Risk Factors That Are Associated with Acceleration of Arterial Ageing
Studies have shown that although ageing is the strongest predictor of arterial stiffness, the degree of arterial stiffness can be accelerated by various risk factors such as hypertension, smoking, high salt intake, dyslipidemia, diabetes mellitus and biomarkers such as high sensitive C-reactive protein (hs-CRP). Among these risk factors, this review article will focus on hypertension, the neurohumoral mechanism and inflammation.
Hypertension
Although age is the most important contributing factor to arterial ageing, its effect is largely influenced by other clinical factors including hypertension, diabetes mellitus, dyslipidemia or vascular inflammation. Benetos et al.16) reported that high blood pressure, high pulse rate and high serum creatinine are independent predictors of arterial stiffness after the analysis of 483 individuals who were examined twice over a 6 years period. In the hypertensive patients, the progression of arterial stiffness measured by PWV change was significantly higher compared to normotensive patients.13) The mechanism could be explained by increased oxidative stress,17) vascular inflammation,18) endothelial dysfunction19) and activation of the renin-angiotensin aldosterone system (RAAS)20) that is characteristic of hypertension. Studies have shown that controlling blood pressure significantly suppresses the progression of arterial stiffness and the suppression of progression regarding arterial stiffness has been demonstrated to be associated with significant improvement in cardiovascular prognosis.21) As the activation of the RAAS pathway is important in the pathogenesis of arterial ageing, antihypertensive drugs that block RAAS may be more effective in reducing arterial stiffness beyond the blood pressure lowering effect. This has been demonstrated in several, small scale studies. Takami et al.22) studied the change of brachial-ankle PWV (baPWV) at the baseline and 3 months after drug treatment with 80 mg of valsartan (angiotensin receptor blocker, ARB), 2-4 mg of temocapril (angiotensin converting enzyme inhibitor, ACEI), 10 mg cilnidipine (L, N type calcium channel blocker, CCB) or 20 mg of Nifedipine (L type dihydropyridine CCB) by the random allocation of 76 patients older than 65 years of age. Interestingly, valsartan, temocapril, and cilnidpine reduced the baPWV value considerably whereas nifedipine hardly did even though there was no difference in the change of blood pressure between the groups (Fig. 1). Among the anti-hypertensive drugs, valsartan demonstrated the strongest effect in reducing the PWV.
Similarly, Munakata et al.23) randomly assigned 41 hypertensive patients into two groups and treated them with valsartan or nifedipine for 3 months. The reduction of the PWV value was only observed in the valsartan group whereas there was no difference in the change of blood pressure between the groups. Both studies commented that nifedipine, which is dihydropyridine CCB, did not reduce the PWV because the drug increased heart rate which is directly proportional to PWV. The limitation of both studies was the fact that as the PWV value was measured just 3 months after treatment, the reduction of PWV may be due to passive destiffening due to blood pressure decrease rather than a long term benefit on the structure of the arteries. However, the fact that there was a significant difference despite a similar reduction in blood pressure suggests a potential beneficial effect of RAAS blockade for reducing arterial stiffness. Also, recent studies have demonstrated evidences to suggest that, in addition to the blood pressure lowering effect, ARB or ACEI have a protective effect against arterial ageing by anti-oxidation, anti-inflammation and improvement of the endothelial function by RAAS inhibition.24)25) Larger studies that could demonstrate whether or not the differential effect on PWV may affect long term cardiac prognosis is needed.
Renin-angiotensin-aldosterone system
Traditionally, RAAS has been known to affect the arteriole and cause systemic vasoconstriction, but recent studies have revealed that RAAS can also change the structure of large arteries by changing the important composition such as collagen and elastin.26) Also, neurohumoral modification on the large artery causing endothelial cell dysfunction has been known to accelerate arterial ageing.27) Recent evidence suggests that excess aldosterone may have a detrimental effect on arterial ageing. In a study done of 438 hypertensive patients who underwent PWV measurement and assessment of the aldosterone-renin ratio (ARR), ARR of at least 20 with a serum aldosterone level higher than 12 ng/dL had significantly higher PWV values compared to hypertensive patients with an ARR value lower than 20 after adjusting for contributing factors (Fig. 2). Multiple linear regression revealed that aldosterone is significantly associated with PWV.28)
Recent evidence suggests that aldosterone blockade using spironolactone reduces arterial stiffness independent from blood pressure. In a study by Edwards et al.29) 112 chronic kidney disease stage 2 or 3 patients with well controlled blood pressure who were taking RAAS blockade at baseline were administered with either spironolactone 25 mg or a placebo. The follow-up PWV measurement at the 40th week of treatment demonstrated that patients who were administered with spironolactone showed a significant reduction in PWV and augmentation index independent from blood pressure reduction (Fig. 3). Aldosterone, through the activation of the mineralocorticoid receptor, may act to increase arterial inflammation and fibrosis. As such, administration of spironolactone may reduce arterial stiffness and retard arterial ageing through the reduction of arterial inflammation and fibrosis.30)31)
Vascular inflammation
Vascular inflammation is a well-known contributing factor for the pathogenesis of arterial ageing. Necropsy studies done in aged human thoracic aortas demonstrate increased levels of angiotensin converting enzyme, angiotensin II, angiontensin receptor type 1 MMPs and MCP-1 compared to the young aortas, suggesting the likelihood for the significant role of inflammation in the pathogenesis of vascular stiffening.32) The activation of the MCP-1/CCR2 pathway, which has been demonstrated to have a significant role in mediating vascular inflammation and remodeling, further stimulates vascular inflammation, increases the expression of cell adhesion molecules, increases the secretion of MMP, amplifies the activity of other cytokines and increases VSMC migration.33-35) Also, studies have demonstrated that inflammatory cytokines stimulate the local production of C reactive protein by VSMCs.36)37) CRP, which is well known to be a marker of systemic inflammation, has been demonstrated to have an active role in promoting vascular inflammation and reducing endothelial function.38-40) CRP has been shown to be associated with endothelial dysfunction, increased cytokine expression, such as monocyte chemoattractant protein-1, and endothelial cell adhesion molecules.41) Recent studies have demonstrated a significant association of hs-CRP and arterial stiffness.42)43) A study by Chae et al.44) demonstrated that in a normotensive population, increase in hs-CRP is associated with the future development of hypertension, suggesting a role for systemic inflammation in the pathogenesis of vascular remodeling and hypertension.
The importance of inflammation in the pathogenesis of arterial ageing can be readily seen in patients suffering from chronic inflammatory disease such as rheumatoid arthritis. Studies have clearly demonstrated that patients with chronic inflammatory disease have increased PWV compared to control subjects in the similar age group and that patients with systemic inflammatory disease have accelerated arterial ageing that is independent from age and blood pressure.45-47) In a study by Galarraga et al.48) twenty six rheumatoid arthritis patients treated with tumor necrosis factor alpha antagonist showed significant attenuation of the augmentation index after 2 and 4 months of treatment which were not observed in 122 patients treated with conventional disease-modifying anti-rheumatic drug regimens. This demonstrates that reduction of the systemic inflammatory process may reduce inflammation in the arteries and subsequently retard the progression of arterial ageing. From the studies that have been done regarding the pathogenesis of arterial ageing, we propose that risk factors such as ageing, hypertension, dyslipidemia, and diabetes all act through the common pathway of increased oxidative stress and vascular inflammation that leads to adverse vascular remodeling and accelerated arterial ageing.
Molecular Mechanism of Arterial Ageing
A disease associated with a proven genetic defect provides us with insight into the pathogenesis, molecular mechanism and possible therapeutic target of the disease. Arterial ageing exhibits such a disease in the Hutchinson Gilford Progeria syndrome (HGPS). The molecular defect caused by a single gene defect in HGPS is well defined and provides numerous of clues for research on the arterial ageing process. HGPS is caused by a point mutation in position 1824 of the LMNA gene, replacing cytosine with thymin, resulting in the subsequent deletion of 150 base pairs in Exon 11 which is an important target sequence of the excision enzyme Zmpste24/FACE1. As a result, the farnesylated C terminus cannot be removed from the prelamin and the protein, which is a major structural component of the nuclear lamina along with lamin C, cannot be integrated into the nucleus (Fig. 4).49) This will eventually result in nuclear disruption, epigenetic and genetic dysregulation, dysfunction of VSMC proliferation and increased apoptosis which will result in accelerated atherosclerosis and premature ageing. For reasons unknown, the aging process is most affected in the VSMC, resulting in the affected patients dying at an average age of 13 years, mostly due to cerebrovascular disease or myocardial infarct.50) Although progerin does not accumulate in the normal ageing process, as oxidative stress increases the expression of Zmpste24/FACE1 which excises the farnesylated C terminus from the prelamin A protein decreases.51) The resultant prelamin accumulation and lamin disruption results in pathologic findings in the vasculature that are similar to those of HGPS.51)52) The results from this study show that any process that increases oxidative stress, such as the clinical risk factors of accelerated arterial ageing, may act to inhibit the expression of Zmpste 24/FACE1 and promote arterial ageing. The farnesyl transferase inhibitor and geranyl-geranyl transferase inhibitor are a class of drugs that inhibit the prenylation of the C terminus of prelamin and studies have shown that the administration of these drugs suppress vascular ageing in-vivo.53) Currently, clinical trials are ongoing to demonstrate the efficacy of the farnesyl transferase inhibitor in patients with HGP syndrome.53-55) A well-known pleiotropic effect of HMG-CoA reductase inhibitors is the inhibition of the synthesis of farnesyl pyrophosphate or geranyl-geranyl pyrophosphate which is an important substrate for progerin formation.56) Some studies reported that statins suppress the progression of arterial stiffening in humans.56)57) More recently, rapamycin, which is an inhibitor of the mammalian target of rapamycin, reportedly may enhance the clearance of mutated lamin by the autophage mechanism (Fig. 5).58) Although rapamycin itself may be too toxic to administer systematically, it provides an attractive target to explore for discovering the possible therapeutic target of arterial ageing.
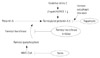
In summary, although arterial ageing is naturally observed in the normal ageing process, various conditions including hypertension, dyslipidemia, diabetes mellitus, cytokines, the genetic condition or inflammation enhance oxidative stress and consequent cellular senescence, and telomere shortening or a lamin A dependent nuclear defect ultimately accelerate the arterial aging process (Fig. 6).
Conclusion
Arterial ageing can be described as age related medial degeneration and sclerosis. Aged arteries are associated with endothelial dysfunction, intima thickening, elastin degradation of the media layer and accumulation of collagen and proteoglycans in the media layer. The increase in the stiffness of the large arteries will result in increased systolic blood pressure and is independently associated with an increased risk of cardiovascular disease. Various conditions that enhance oxidative stress and cellular senescence, through the inhibition of the expression of Zmpste24/FACE1, cause the accumulation of prelamin and nuclear defects which will accelerate the arterial ageing processes. Recently, in vivo studies regarding the efficacy of drugs to reduce the laminopathy such as the farnesyl transferase inhibitor or rapamycin have been promising and provide new insight into the mechanism and treatment of arterial ageing in the future.





 XML Download
XML Download