

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
An angiotensin-II receptor blocker (ARB) is a representative antihypertensive drug. It is also effective in left ventricular dysfunction, has a reno-protective effect reducing proteinuria, and an atrial reverse-remodeling effect as an electrophysiologic stabilizer.1) Studies of the reverse-remodeling effect are relatively rare, and some investigators do not agree on some points of view. Atrial fibrillation (AF) has a relationship with atrial remodeling. Angiotensin converting enzyme lowers bradykinin, which increases fibrosis and collagen deposition in atrial tissue.2) Goette et al.3) explained that the decrease of bradykinin is due to the angiotensin converting enzyme dependent extracellular signal-regulated kinases (Erk1/Erk2). An activated angiotensin-II receptor activates mitogen-activated protein kinase. As a result of histologic change, atrial enlargement occurs and the atrium may be a substrate of AF.4) The other mechanism of atrial remodeling is electrical remodeling. Angiotensin activation induces myocyte calcium overload, which presents a prolongation of the refractory period, depolarization-delay, and the increase of automaticity. Finally, it makes substrates for AF. Similarly, the renin-angiotensin-aldosterone system has a relationship with atrial remodeling, which has a relationship with AF development and maintenance.
It is widely understood that hemodynamic overload in the atrium is one of the most important factors in atrial fibrosis5) and that structural changes such as atrial enlargement and fibrosis have a relationship with atrial dysfunction.6)7) Li et al.7) reported that electrical in-homogeneity due to atrial fibrosis plays an important role in AF induction and maintenance in a canine heart failure model. Gap junction builds signal propagation channels to neighboring myocytes. Its geometrical distortion can form an in-homogeneous electrophysiologic network, and then, the consolidation of AF. These histological changes go with atrial fibrosis and the expression of connexin 43 and connexin 40 proteins.8) However, those results showed wide variations, with even opposing results within the same models. Therefore, those results cannot define the definite causal-relationship between arrhythmia and connexin.9)
Losartan may prevent left ventricular systolic dysfunction in a rat myocardial infarction model.10) The Renin-Angiotensin-Aldosterone system has a relationship with the pathomechanism of AF. Kumagai et al.1) reported that angiotensin receptor blockers can prevent atrial electrical remodeling in the canine model, in which he used rapid atrial pacing to induce AF. In the heart failure model, we conducted this study to evaluate the reverse remodeling effect of the angiotensin receptor blocker, showing echocardiographic findings, the expression of cardiac connexin, and AF inducibility.
Subjects and Methods
Experimental animals and substance
For the experiment, male Sprague-Dawley rats (Jung-Ang Animal, Korea) of around 260 g in weight were used. Each rat was isolated and bred in an individually ventilated microisolator cage rack system, in which day and night were set at 12 hours each. The rats were fed with standard rodent provender and distilled water. This experiment was performed, abiding by the guideline of the Chonnam University Hospital animal subject institutional review board. We estimated the amount of provender for each subject for 5 days of adaptation. The grinded medicine was mixed up into grinded provender, which was congealed for one day at a temperature of 35℃.
We used 25 rats, divided into the sham group (n=5), the heart failure group (n=10), and the heart failure-ARB group (n=10). Treatment was performed with the ARB, Cozaar® (losartan kalium, Merk Sharp & Dohme Ltd, UK). 30 mg/kg of losartan kalium was placed on each of the heart failure-ARB groups for 4 weeks.10)
Heart failure model
The ischemic heart failure model was induced with methods that Johns and Olson11) presented. Anesthesia was induced with a ketamine (50 mg/kg) and xylazine (5 mg/kg) intramuscular injection. Artificial ventilation was performed with a small animal mechanical ventilator (Harvard Apparatus, Holliston, MA, USA) after tracheal intubation. The rat heart was exposed after a left thoracotomy in the supine position. Myocardial infarction was obtained via ligation of a left anterior descending coronary artery in the heart failure and heart failure-ARB groups. The chest was closed immediately. In the sham group, only a thoracotomy and closure were performed. The rats were bred for 4 weeks.
Echocardiography
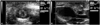
Echocardiography was performed under anesthesia with a ketamine (50 mg/kg) and xylazine (5 mg/kg) intramuscular injection prior to the operation and 4 weeks after the operation. A 15 MHz linear prove (Acuson-Siemens Sequoia C512, Mountain View, CA, USA) was used to perform the transthoracic echocardiography. Left ventricular end systolic diameter, left ventricular end diastolic diameter, left atrial diameter (LAD) in the parasternal long axis view, and left ventricular ejection fraction (LVEF) were measured (Fig. 1). All echocardiograpies were performed in a blind state.
Induction of atrial fibrillation
After the follow-up of echocardiography, AF induction was performed under anesthesia. After 10 minutes of an equilibration period following anesthesia, a 4 Fr electrode catheter (St. Jude Medical, St Paul, MN, USA) was inserted into the esophagus and positioned to ensure constant atrial capture. A BLOOM 215B DTU stimulator, Prucka recording system, Grass S-8800 2 channel stimulator (Grasstech, Quincy, MA, USA), MP150, ECG100C ×2 (Biopac, Santa Barbara, CA, USA) were used for stimulation and recording. Atrial pacing was performed at twice the diastolic threshold (10 volts, 500 ohm, and 20 mA), using 2 poles on the pacing catheter (pulse width: 5 milliseconds). To induce AF, the burst stimulation at a frequency of 25 millisecond intervals was performed during 35 seconds in each of the rats (Fig. 2).
Histologic analysis
After the AF induction test, all subjects were ceased. For 5 of 8 rats in each group, the hearts were fixed by 10% formaldehyde perfusion into the abdominal aorta. After formaldehyde perfusion, the hearts were excised immediately. The excised hearts were fixed in 10% formaldehyde. Hematoxylin-eosin stain, Masson's trichrome stain, and immunohistochemical stain for connexin 43 (rabbit polyclonal anti-Cx43 antibody, 1/100, Zymed-laboratories, CliniSciences, Montrouge, France) were performed. Fibrosis and connexin expression were quantified by automatic computer graphic software (Image J®, Adobe® Photoshop® Cs3®).
Western blot analysis
For the Western blot analysis of connexin 43, from 3 of 8 rats, both atria were excised after sacrifice with CO2 inspiration. Western blot analysis was performed on lysates from frozen tissue. A 1% NP buffer was used as the lysis butter. 50 µg of protein extracts were used. Connexin 43 was detected using rabbit polyclonal anti-Cx43 antibody (1/100, Zymed-laboratories).
Statistical analysis
Statistical analysis was performed using computer software Statistical Package for the Social Sciences (SPSS) 15.0 for Windows® (SPSS Inc., Chicago, IL, USA). The Mann-Whitney test was used to compare groups. Intra-group analysis was performed by a paired t-test. Statistical significance was defined when the p was less than 0.05.
Results
Heart failure model
Groups were divided into the sham group (n=5), heart failure group (n=10), and heart failure-ARB group (n=10). After LAD ligation, 10 rats expired. We maintained 5 rats in each group for 4 weeks after operation. Weight gain did not differ between the heart failure group and the heart failure-ARB group for 4 weeks (Fig. 3A).
Echocardiography
Baseline LVEFs in the sham, heart failure group, and heart failure-ARB group were 54.4±4.07%, 67.8±10.2%, and 60.7±6.43%, respectively. Follow-up LVEFs were 58.7±2.36%, 44.3±6.23%, and 47.5±11.74%, respectively. The decrease of LVEF in the heart failure-ARB group was less than that in the heart failure group (p=0.023) (Fig. 3B). Baseline LADs in the sham, heart failure group, and heart failure-ARB group were 3.9±0.32 mm, 4.2±0.12 mm, and 4.6±0.53 mm, respectively. Follow-up LADs were 5.5±0.75 mm, 8.5±0.44 mm, and 6.5±0.67 mm, respectively. The increase of LAD in the heart failure-ARB group was less than that in the heart failure group (p=0.025) (Fig. 3C). The size of the left atrial appendage in the heart failure-ARB group was less than that in the heart failure group.
Atrial fibrillation inducibility
The inducibility of AF in the sham, heart failure group, and heart failure-ARB groups were tested. There was a trend of high AF inducibility in the heart failure group. However, the inducibility was not steady.
Histology, immunohistochemical stain, Western blot
Grossly speaking, the size of the excised heart in the heart failure group and the heart failure-ARB group was larger than that in the sham group. The area of infarction was pale and thin (Fig. 4).
On the hematoxylin and eosin (H&E) stain (Fig. 4), in the heart failure group, definite dilatation of the atria and ventricles was verified. Marked thinning of the wall in the infarcted area was verified. The H&E stain showed the destructive feature of the myocardial contraction unit, abnormal sarcomere, and definite interstitial fibrosis. In contrast, atrial myocyte preserved a relatively intact myocardial contraction unit and normal sarcomere in the heart failure-ARB group (Fig. 5).
The Masson's trichrome stain revealed severe fibrosis of the infarcted ventricular area and more fibrosis of the atria in the heart failure group (Fig. 5). The left atrial appendage area of fibrosis measured by Masson's trichrome stain in the heart failure group and the heart failure-ARB group were 2.192% and 0.744%, respectively. The left atrial fibrosis area of the body measured through the Masson's trichrome stain in the heart failure group and the heart failure-ARB group were 8.113% and 4.311%, respectively (Fig. 5).
The immunohistochemical stain for connexin 43 (rabbit polyclonal anti-Cx43 antibody, 1/100, Zymed-laboratories, CliniSciences, France) showed a significant amount of plasmalemmal distribution of connexin 43 in the heart failure group. Connexin 43 staining was not seen on the intercalated disc (Fig. 6B). In the heart failure-ARB group, some slight connexin 43 protein staining was seen on the intercalated disc (Fig. 6C).
Western blot of the connexin 43 protein showed that the heart failure group had stronger connexin 43 bands than those of the sham group, and the heart failure-ARB group had weaker connexin 43 bands than those of the heart failure group (Fig. 6).
Discussion
Atrial fibrillation has a relationship with atrial remodeling. It is relatively common to observe arrhythmia, which can be paroxysmal and persistent. It can develop with or without underlying heart diseases. It has clinical significance because of probable hemodynamic instabilities and thromboembolic events. The multiple-wavelet hypothesis has been an accepted theory as a mechanism of AF, which is that multiple wavelets conflict and collide, and then form small wavelets repeatedly.12)
Atrial fibrillation is induced by initial abnormal electrical activity and is maintained in the atrium with irregular reentry. These irregular electrical reentries are related with structural and electrical remodeling. Researchers have studied an ion channel protein like connexin, which seems to play an important role in the remodeling.13) The remodeling is frequent in myocardial infarction. The reason as to why AF develops frequently in old ventricular myocardial infarction is not widely or thoroughly understood.14) The current understanding is that old ventricular myocardial infarction forms atrial structural and electrical remodeling, which serve substrates for AF. Old ventricular myocardial infarction may induce electrical in-homogeneity of the atrium and sympathetic over-distribution in the atrium without atrial infarction. These conditions seem to be sufficient to induce AF.14) From these points of view, the rat ischemic heart failure model was induced by myocardial infarction, formed by left coronary artery ligation in this experiment. Therefore, we were able to form a similar substrate with theoretical conditions for AF maintenance. We observed improved LVEF and a smaller increase in the LAD in the heart failure-ARB group. These findings suggest the preventive effects of ARB on atrial structural remodeling. In human beings, dilatation of an infracted ventricle develops with hypertrophy of non-infarcted myocardium over months or years.15) In this experiment, the thinning of the infarcted area and the hypertrophy of the non-infarcted area developed in 28 days. These rapid changes seem to be due to the high left ventricular systolic pressure, like that of human beings, and the difference of myocardial thickness between humans and rats. We do concede limitations in echocardiographic comparison. It would be better to compare the immediate post-OP and later echocardiogram. However, we did conduct a pre-OP echocardiogram to reduce stressful situations and procedure times.
Atrial systolic function might not recover even though cardioversion is performed chemically or electrically. We think that the cause of this phenomenon is that structural and electrical remodeling of the atrium cannot be reversed quickly. The fact that persistent AF can induce chronic hibernation-like change in atrial myocardium may explain those slow reversals.6)
In this study, H&E stain and Masson's trichrome stain revealed less fibrosis in the heart failure-ARB group. These findings suggest the preventive effects of ARB on atrial structural remodeling. In-homogeneity of the gap junction is one of the structural remodeling activities in chronic AF. We think that gap junction in-homogeneity is the result of atrial dilatation and AF itself. In other words, AF begets AF, meaning that atrial dilatation and fibrillation can be induced in ventricular failure. Remodeling of the gap junction may change the conduction properties and then contribute to the maintenance of AF. It is not clear how important the role remodeling plays in the induction of AF.13) We propose that gap junction remodeling may reduce conduction velocity in the atrium.
Thomas et al.16) reported that a 50% decrease of ventricular conduction velocity was observed with a decrease of connexin 43, indicating that connexin 43 played a significant role in electrical conduction between ventricular myocytes. In contrast, the electrical conduction velocity in the atrium was not affected by the decrease of connexin 43, but connexin 40. It was suggested that connexin 43 is ventricular-specific and connexin 40 is atrial-specific. However, after their report, it has been recognized that both connexin 40 and connexin 43 are related with AF.
In this study, connexin 43 showed a significant amount of plasmalemmal distribution in the heart failure group and less of a connexin 43-stained area in the heart failure-ARB group. Furthermore, Western blot analysis showed that the heart failure group had stronger connexin 43 bands than those of the heart failure-ARB group. We thought that the change of quantity and distribution of connexin 43 was the result of the change in dephosphorylated connexin 43 in the state of myocardial infarction or ischemic heart failure. It would be better to compare these in the same phosphorylation condition. Unfortunately, we could not standardize or equalize the phosphorylation.
Some studies reported changes of connexin quantity or arrangement in some species by species.17) The results were quite varied. Some reported opposite results even in the same model. We regard this range of results as a result of different species. Therefore, we reviewed several studies using a rat model. Hoyano et al.18) reported the down-regulation of connexin 43 in an autoimmune myocarditis model. Reil et al.19) reported no change of connexin 43 in an aldosterone infusion model. Rucker-Martin et al.20) reported the up-regulation of connexin 43 in a heart failure model.
The result of this study is compatible with the results of Rucker-Martin et al.20) Regardless of the connexin result, for the bedside physician, what is important is whether the ARB has the potential for upstream therapy in AF or not. Unfortunately, in our induction study of AF, we could not verify the difference between the heart failure and the heart failure-ARB groups. Some researchers reported that candesartan can prevent AF, inhibiting atrial structural remodeling.1) In an experiment of the canine model, the inhibition of the angiotensin-II receptor could prevent the shortening of the atrial effective refractory period during rapid atrial pacing. This is solid evidence proving the relationship between angiotensin II and atrial electrical characteristics.21) The 2010 European Society of Cardiology guidelines recommended that angiotensin converting enzyme inhibitors and ARBs should be considered for the prevention of new-onset AF in patients with heart failure and reduced ejection fraction.22) The 2012 European Society of Cardiology guidelines have said that there is now very little reason to consider the use of upstream therapy for the prevention of AF recurrence in patients with little or no underlying heart disease.23)
Finally, the ARB has little preventive effect on AF if there is no underlying heart disease, such as heart failure. However, we do not deny that the ARB has an atrial reverse remodeling effect. We should keep in mind that AF is an outgrowth of complex molecular, mechanical, and electrical remodeling. Thus, we regard gap junction as potentially not being a surrogate target of atrial remodeling.
Generally speaking, the ARB is the recommended medication in AF patients with heart failure or left ventricular systolic dysfunction. However, according to recent guidelines,23) its effect on the prevention of AF is not clear.
In the ischemic heart failure model of rats, structurally and histologically, the ARB, losartan, has atrial reverse-remodeling effects. However, electrically, its role as an electrical stabilizer requires further study.





 XML Download
XML Download