

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Hypertrophic cardiomyopathy (HCMP) is a heart disease instigated by a variety of causes, such as genetic syndromes, e.g., Noonan syndrome and Costello syndrome, metabolic diseases, such as long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency and adenosine triphosphate (ATP) synthase deficiency, familial history, maternal diabetes mellitus, steroid use and etc. These can also be caused by a variety of reasons; heart and other organs are often accompanied by other malformations.1) According to a study performed by Oh et al.,2) incidences of primary HCMP are very rare. As for Korea, the occurrence is 0.51 per hundred thousand people, and if symptomatic, it is known to have a poor prognosis. Here, we describe a neonate with congenital HCMP without other congenital anomalies who was presented with severe congestive heart failure and respiratory failure, and whose condition proceeded rapidly to death during an early neonatal period.
Case
A 2726 g (<50 percentile) male newborn was delivered via cesarean section at 38 weeks' gestation as the first child of healthy, nonconsanguineous parents. His mother (gravida 3, para 2) was 35-year-old and his father was 37-year-old at the time of the delivery. The mother was healthy and her family history was unremarkable for cardiovascular disease or genetic disease, but her first pregnancy ended in stillbirth. No abnormality was detected by ultrasonography during perinatal care. Because of high-risk pregnancy, due to older age of the mother, karyotyping was performed from the amniotic fluid, which the result showed 46 normal chromosomes. Maternal hypertension developed at 38 weeks of pregnancy, which required emergency caesarian section. After birth, the baby was transferred to our neonatal intensive care unit because of dyspnea, cyanosis and generalized hypotonia. The Apgar scores at 1 and 5 minutes after birth were 3 and 7, respectively. The initial vital signs were body temperature 36.0℃, blood pressure 73/54 mm Hg, heart rate 167/min and respiratory rate 69/min. An endotracheal tube was immediately placed because of tachypnea, chest wall retraction and cyanosis. In the physical examination, head circumference was 33.5 cm (<75 percentile) and body length was 50.5 cm (<90 percentile). The baby had no dysmorphic features, including head shape, shape and location of eyes, ears and nose. Chest and abdomen showed no abnormal findings, except chest wall retraction. The heart beat was regular with tachycardia, systolic murmur graded as III was auscultated on the right upper sternal border. Respiratory findings were normal. There was no organomegaly and normal neurologic exam. Laboratory studies of the serum yielded normal results for the following: haematological parameters, electrolytes, liver function tests, renal function, calcium, phosphate, alkaline phosphatase, creatine kinase, cholesterol, triglycerides, glucose, and pyruvic acid. Serum B-type natriuretic peptide was increased at 1610 pg/mL. Blood culture showed no detection of bacteria. The serum Immunoglobulin M levels of Toxoplasma, Rubella, Cytomegalovirus and Herpes simplex virus were negative. An inborn error of metabolism screening test was done and the result was normal. Chest X-ray revealed dextrocardia, increased pulmonary vascular markings and cardiomegaly. The abdomen X-ray revealed normal location of the liver, spleen and intestine. The abdominal sonography showed no dysmorphic features of abdominal organs. Electrocardiogram showed sinus rhytm and tarchycardia.
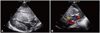
On the first day after birth, two-dimensional echocardiography demonstrated marked hypertrophy of both ventricles (the ventricular septum was 15-mm-thick and the posterior wall of the left ventricle was 12-mm-thick). Ventilator weaning was not possible and the baby required intermittent positive-pressure ventilation. On the 26th day after birth, two-dimensional echocardiography follow-up demonstrated an increment of ventricular septum and posterior wall thickness of the left ventricle, compared to the first day, showing the ventricular septum with 17-mm-thickness and the posterior wall of the left ventricle with 13-mm-thickness (Fig. 1A). The cavities of the left and right ventricle were small due to the hypertrophied ventricular septum and wall. Narrowing of the outflow tracts in the left and right ventricles resulted in obstruction with pressure gradients measured at 45 mm Hg in the left ventricular outflow tract and 35 mm Hg in the right ventricular outflow tract. The hypertrophy of both ventricles had progressed on the 38th day after birth, and the thicknesses of the ventricular septum and posterior wall of the left ventricle had increased to 21 mm and 15 mm, respectively. A color and pulse Doppler echocardiogram also demonstrated an accelerated flow velocity at the outflow tract of the left ventricle (Fig. 1B). As HCMP was aggravated, left ventricular capacity was decreased gradually, and these brought decreased blood pressure, elevated heart rate and also decreased urine output. Further, as resistance of left atrium was increased, pulmonary congestion was also gradually aggravated. He had no arrhythmia and no infection focus. On the 51st day after birth, despite the administration of maximum dose of inotropics, blood pressure decreased consistently. The heart rate also decreased slowly, finally reaching a rate below 60/min. The infant expired during cardiopulmonary resuscitation without response due to heart failure, and respiratory failure due to pulmonary congestion. An autopsy was performed on the following day after death, showing the thickness of the left and right ventricle wall with about 3.0 cm and 2.0 cm, respectively. Additional abnormality of the heart was not detected, except the already known dextrocardia. No specific findings of the other organs were seen. The microscopic findings of the myocardium indicated myofiber hypertrophy with nuclear enlargement and hyperchromasia. The nuclei predominantly showed a typical box shape (Fig. 2).
Discussion
Hypertrophic cardiomyopathy in children may be either primary, due to a mutation in one of several genes,3) or secondary, due to the sequelae of obstructive congenital heart disease or an inborn error in metabolism, such as glycogen storage disease or mucopolysaccharidosis.4) In addition, there have been reports of primary HCMP occurring in association with other congenital heart lesions, such as ventricular septal defects and tetralogy of Fallot.5)6) As mentioned earlier, congenital HCMP can occur in a newborn or fetus due to chromosomal abnormality, mutation of protein metabolism, maternal diabetes or exposure to corticosteroids. According to a study in patients with neonatal cardiomyopathy, performed between 1995-2004 by Badertscher et al.,1) among a total of 14 patients with HCMP, 7 cases had genetic causes, such as Noonan syndrome, Costello syndrome, Cantù syndrome and etc., 2 cases had a metabolic disease in form of ATP synthase deficiency, 1 case experienced myopathy, and 4 cases were categorized as unexplained causes. The results of a follow-up until 1 year after birth showed that 4 patients (29%) had died during that period. In Korea, several cases were reported with congenital HCMP accompanied by multiple anomalies, due to chromosomal abnormality, such as Noonan syndrome, Leopard syndrome or Costello syndrome.7-9) In our case, the infant had no family history of HCMP, his mother was not diabetic, his karyotype was normal, and he had no history of exposure to corticosteroids or showed any inborn errors in the metabolism. Moreover, he had no other congenital heart disease that could have resulted in HCMP. We considered performing a septal myectomy or transplantation, despite the small chance of success. However, the patient's myocardium showed aggravation with rapid thickening, and he ultimately died from congestive heart failure and respiratory failure, due to pulmonary congestion. Recent genetic studies of sporadic or familial HCMP have identified a variety of mutations localized to chromosomes 1, 11, 14, and 15, and the mutations in genes encoding sarcomere proteins are autosomal dominant.4)5) HCMP is one of the more common heritable cardiac diseases, and thus, further genetic studies of congenital HCMP are warranted.

 XML Download
XML Download