

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Doxorubicin is a glycosidic anthracycline anticancer drug, and it was first isolated from Streptomyces peucetius in 1960s. Although it has a strong anticancer effect, doxorubicin is also known to cause cardiotoxicity that leads to arrhythmia and congestive heart failure.1) A variety of studies have been tried to discover the mechanisms involved in doxorubicin-induced cardiotoxicity, and apoptosis induced by reactive oxygen species has been inferred as the most important mechanism to explain doxorubicin-induced cardiotoxicity.2)3) Therefore, understanding the doxorubicin-induced apoptosis signaling process and investigation of the possible candidate molecules which have an anti-apoptotic effect against doxorubicin injury are crucial.
Survivin is the smallest member of the inhibitor of apoptosis (IAP) gene family and it is known as a bifunctional protein that acts as an apoptosis suppressor, and is an important factor in regulating proper cell division.4) Although its mechanism is not clearly understood, survivin is considered to be a key factor in regulating cell survival and suppression of apoptosis. Survivin is overexpressed during development and in most human cancer tissues. It has been reported that survivin was able to suppress caspase-3 and -7 even though the in vitro binding experiment failed to show this result. It is also known that survivin interacts with Smac, which is the inhibitor of other IAPs.5) Such accumulated evidence suggests that survivin has the potential to regulate apoptosis and cell survival. Survivin is also detectable in terminally differentiated tissues such as the myocardium.6) However, the molecular function of survivin in cardiac myocytes has not been clearly determined.
Since the main role of survivin is considered to be the suppression of caspases and apoptosis, survivin may be one of appropriate targets for the doxorubicin-induced cardiac myocyte apoptosis signal cascade. Apoptosis of cardiac myocytes leading to loss of contractile units has been implicated in the development of cardiomyopathy.7) The microtubule localization of survivin and the enhancement of microtubule structure stability with the overexpression of survivin are important evidence to show that survivin might be one of the targets for doxorubicin injury because those cytoskeletal proteins are essential in heart contraction.8) Furthermore, it was reported that the inhibition of survivin resulted in similar biochemical interactions as the treatment of doxorubicin9) and heart failure was induced in a survivin knock-out mouse model.10)
In the present study, a recombinant survivin that was fused to the protein transduction domain (PTD) derived from HIV-TAT protein11) was delivered into H9c2 cardiomyocytes. A viral carrier was used because the transfection efficacy of delivery to cardiac myocytes with a non-viral carrier is extremely poor. And then, we evaluated the anti-apoptotic effect of PTD-mediated transduction of the recombinant survivin against doxorubicin injury with the apoptosis-related signals.
Materials and Methods
Construction and protein purification of TAT-survivin
The survivin fragment was inserted into the BamHI and XhoI sites of the pHis/TAT vector12) for TAT-survivin fusion protein expression. E. coli BL21 transformed with recombinant plasmid was expressed at 37℃ in Lunia-Bertani broth. The bacterial pellet was harvested and resuspended in buffer (8 M urea, 100 mM NaCl, and 20 mM Hepes, pH 8.0). The clarified lysate was loaded onto a Ni-iminodiacetic acid affinity column (Macherey-Nagel, Germany). TAT-survivin protein was eluted with imidazole in buffer. The proteins were loaded onto a PD-10 desalting column in order to exchange the buffer to phosphate buffer saline (PBS).
Cell culture and protein transduction into cells
The rat heart-derived myoblast cell line, H9c2 cardiac myocytes, were obtained from the American Type Culture Collection (USA). H9c2 cardiac myocytes were cultured in Dulbecco's modified Eagle's Medium (DMEM) supplement with 10% fetal bovine serum (FBS) and 1% penicillin streptomycin (Gibco, USA) at 37℃ in humidified atmosphere of 5% CO2. All experiments were performed using cells between 10 to 25 passage numbers. H9c2 cardiac myocytes were incubated for 24 hours in 100 mm culture plate and changed to 0.5% FBS DMEM for 24 hours starvation. After starvation, they were treated with 1 µM of doxorubicin (Tocris, USA) for 24 hours. For the transduction of TAT-survivin, cells were treated with 1 µM of TAT-survivin for 1 hour prior to doxorubicin.
Subcellular fractionation
Cell pellets were separated into cytoplasmic, nucleic, membrane and mitochondrial fractions using the Mitochondria Isolation Kit (Qiagen, Germany) according to the manufacturer's instructions. After washing, cells were suspended in lysis buffer to disrupt the plasma membrane without solubilizing it and to aid in the isolation of cytosolic proteins. Plasma membranes and compartmentalized organelles, such as nuclei, mitochondria, and the endoplasmic reticulum, remained intact and were pelleted by centrifugation at 1000×g for 10 minutes. The resulting pellet was resuspended in disruption buffer, repeatedly passed through a narrow-gauge needle (26 or 21 gauge), and re-centrifuged to pellet nuclei, cell debris, and unbroken cells at 1000×g for 10 minutes. The supernatant which contained mitochondria and the microsomal fraction was recentrifuged to pellet mitochondria at 6000×g for 10 minutes. After removal of the supernatant, mitochondria and nuclei were dissolved using a lysis buffer and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis as outlined below.
Reverse transcription-polymerase chain reaction
Total ribonucleic acid (RNA) isolated from cells using QIAzol-Reagent (Qiagen, Germany) was reverse transcribed using Omniscript Reverse Transcriptase (Qiagen, Germany). The cDNAs was amplified using Taq deoxyribonucleic acid (DNA) polymerase (Cosmo Genetech, Korea). The following primer sequences were used: Bcl-2 primers 5'-GACGCGAAGTGCTATTGGT-3' and 5'-TCAGGCTGGAAG GAGAAGAT-3', GAPDH primers 5'-AATGCATCCTGCACCACCAACTGC-3' and 5'-GGAGGCCATGTAGGCCATGAGGTC-3'. Polymerase chain reaction (PCR) products were separated by electrophoresis in a 1% agarose gel containing Gel-red (Biotium, USA).
Immunoblot analysis
Cells were solubilized in a cell lysis buffer (Cell signaling, USA) and centrifuged at 13000 rpm for 1 hour at 4℃. The proteins samples were separated by an SDS-polyacrylamide gel and transferred to polyvinylidene difluoride membranes. After blocking in TBS-tween 20 (TBS-T, 0.1% tween 20) containing 5% non-fat dry milk, the membrane was treated with the appropriate primary antibodies: survivin, cleaved caspase-3, phospho-cyclic adenosine monophosphate response elements-binding protein (CREB) (Ser133), CREB, phospho-p38MAPK (Thr183/Tyr185), p38MAPK (Cell Signaling, USA), BAX, Bcl-2 (Abcam, USA), Smac, β-actin, GAPDH, Lamin B, His-tag (SantaCruz Biotechnology, USA), cytochrome C (Clontech, USA), followed by incubation with the appropriate horseradish peroxidase-sconjugated secondary antibodies. The antigen-antibody bands were detected using enhanced chemiluminescence reagent kit (Millipore, USA) and quantified by densitometry.
Cell viability, caspase-3 activity and terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling assay
Cell viability was measured by the classical 2-(4,5-dimethyltriazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Amresco, USA). H9c2 cardiac myocytes were incubated for 24 hours in 24-well plate and changed to 0.5% FBS DMEM media for 20 hours starvation. Cells were pretreated with TAT-survivin 1 µM for 1 hour, and then treated with 1 µM of doxorubicin for 24 hours. Cell viability was determined by MTT solution which was added to each well to a final concentration of 0.5 mg/mL and was incubated at 37℃ for 1-2 hours. The formazan crystals were dissolved by adding dimethylsulfoxide and absorbance was measured at the 570 nm with a spectrophotometer.
The activity of caspase-3 in the cells was determined spectrophotometrically with an Apoalert™CPP32/caspase-3 assay kit (BD Biosciences, USA) by measuring the release of the chromophore, p-nitroanilide (pNA), following hydrolysis of DEVD-pNA. Terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling (TUNEL) analysis was performed with a commercially available kit according to the manufacturer's instructions (Intergen, USA).
Immunofluorescence microscopy
The cells were incubated on Lab-Tek chamber slides (Nalgene Nunc, USA) and treated with 1 µM wt-survivin or TAT-survivin. Following incubation for 1 hour, the cells were washed twice with PBS and then fixed with 3% paraformaldehyde for 10 minutes at room temperature and washed with PBS. The cells were permeabilized in 0.5% Triton X-100 buffer (0.5% Triton X-100, 20 mM Hepes-KOH, pH 7.9, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose) in PBS for 10 minutes and washed with PBS. They were blocked with PBS containing 0.3% goat serum and 5% bovine serum albumin for 1 hour at room temperature and then incubated for 1 hour with the His-tag antibody. The cells were washed with PBS once and incubated with rhodamine Red-X, goat anti-rabbit IgG (R6394, dilution 1 : 500; Invitrogen) as secondary antibody for 1 hour in a dark room. After washing, the cells were mounted with ProLongantifade reagent containing 4'-6-diamidino-2-phenylindole. The immunoreactive signals were visualized by confocal laser scanning microscope LSM700 (Carl Zeiss, Germany).
Results
Doxorubicin decreases survivin level in H9c2 cardiac myocytes
When the cells were treated with various amounts of doxorubicin for 24 hours, cell viability gradually decreased to about 60% with up to 1 µM of doxorubicin (p<0.05) (Fig. 1A). The treatment of 5 µM of doxorubicin did not further decrease cell viability compared to that of 1 µM of doxorubicin. We next studied whether the expression level of survivin was altered by doxorubicin treatment. The expression of survivin was dramatically suppressed when the cells were treated with no more than 1 µM of doxorubicin (Fig. 1B). Therefore, we fixed the amount of doxorubicin to treat H9c2 cardiac myocytes as 1 µM for further experiments. And Fig. 1C shows the time-dependent decrease of survivin following treatment with 1 µM of doxorubicin.
TAT-survivin fusion protein is efficiently delivered into H9c2 cardiac myocytes
TAT-survivin protein was transduced into H9c2 cardiac myocytes efficiently in a concentration-dependent manner (data not shown). TAT-survivin (1 µM) was detected by immunoblot using anti-His-tag antibody (Fig. 2A). And the immunofluorescence microscopy image using primary anti-His-tag antibody and Rhodamine-conjugated secondary antibody also showed efficient transduction of TAT-survivin protein (Fig. 2B). The red and blue staining in the image represent the transduced TAT-survivin proteins and nuclei, respectively.
TAT-survivin protein protects H9c2 cardiac myocytes from doxorubicin-induced apoptosis
We next examined whether intracellular delivery of TAT-survivin protected H9c2 cardiac myocytes against doxorubicin-induced injury. We observed a significant protective effect of the TAT-survivin protein against doxorubicin-induced cell death. As shown in Fig. 3A, caspase-3 activity was increased more than 3-fold after doxorubicin treatment. However, TAT-survivin transduction significantly decreased caspase-3 activity (p<0.05) (Fig. 3B). Also, TUNEL assay demonstrated the anti-apoptotic effect of TAT-survivin against doxorubicin-induced injury (p<0.05) (Fig. 3C).
TAT-survivin transduction leads to recovery of expression of Bcl-2 but not Bax
Apoptotic signals were evaluated to define the effect of TAT-survivin transduction on doxorubicin-induced apoptosis. To get information about the relationship between doxorubicin-induced apoptosis and apoptosis related signals, we first measured the changes of Bax and Bcl-2 by immunoblot analysis. As shown in Fig. 4A, Bax protein level was increased following doxorubicin treatment, whereas Bcl-2 level was decreased. Treatment of TAT-survivin protein led to significant recovery of Bcl-2 in doxorubicin stimulus. However, there was no change observed in Bax protein with TAT-survivin treatment under the same conditions (Fig. 4A). Reverse transcription-PCR showed that the expression of Bcl-2 was down-regulated by doxorubicin treatment, which was significantly recovered by TAT-survivin transduction (Fig. 4B). Next, we investigated the subcellular localization of a proapoptotic mitochondrial protein Smac and cytochrome C, endogenous inhibitors of IAPs, released from mitochondria during apoptosis. TAT-survivin protein also inhibited the release of Smac and cytochrome C from the mitochondria to the cytoplasm induced by doxorubicin treatment (Fig. 4C). The activation of CREB, which is known as a transcription factor of Bcl-2, was quantified in both whole cell lysate and nuclear protein. As shown in Fig. 4D, doxorubicin prevented the nuclear translocation of CREB. However, a significant restoration of CREB nuclear translocation was observed in the treatment of TAT-survivin protein (Fig. 4D). The phosphorylated form of p38 MAP kinase, which is known as the upstream signal regulator of doxorubicin-induced apoptosis was decreased with TAT-survivin treatment (Fig. 4E).
Discussion
Doxorubicin is one of the most widely used anticancer drugs, but its cumulative and dose-dependent cardiotoxicity has been a major concern in clinical practice for decades. This doxorubicin-induced cardiotoxicity leads to cardiac myocyte death, progressive cardiomyopathy, and congestive heart failure.1) Although intensive investigations on doxorubicin-induced cardiotoxicity have been performed over the decades, the underlying mechanisms responsible for doxorubicin-induced cardiotoxicity have not been completely elucidated. A rapidly expanding body of evidence supports the notion that cardiac myocyte death by apoptosis and necrosis is a primary mechanism of doxorubicin-induced cardiotoxicity.1-3)
In the present study, survivin, known as a strong anti-apoptosis factor, was successfully delivered into H9c2 cardiac myocytes using the PTD system, leading to a significant cytoprotective effect against doxorubicin-induced apoptosis. The transduction of TAT-survivin significantly reduced cell death and caspase activity. The induction of Bcl-2, which is known as an anti-apoptosis factor, also manifested the anti-apoptotic effect of TAT-survivin. Both protein level and mRNA level of Bcl-2 were increased by TAT-survivin transduction. We also demonstrated that the activation of CREB, which is the transcription factor of Bcl-2, was also increased by TAT-survivin. CREB has been established as a cellular transcription factor which binds to a specific DNA site called cyclic adenosine monophosphate response element (CRE) that regulates the transcription of several proteins including the expression of the IAP family,13)14) leading to an upregulation of Bcl-2 expression that is related to anti-apoptosis. Other reports showed that activation of CREB also increased the expression level of the IAP family.15) The present findings, with protein delivery of TAT-survivin, suggest that Bcl-2 may be at least in part involved in the cytoprotection mechanism with TAT-survivin against doxorubicin insult, and that recovery of Bcl-2 level may be attributed to the CREB reactivation by TAT-survivin treatment. Furthermore, TAT-survivin transduction recovered its translocation of Smac into mitochondria from the cytoplasm. Smac is a mitochondrial protein that promotes apoptosis by inhibiting IAPs. Survivin is considered to inhibit Smac by binding to it in the cytosol,5) but the transduction of survivin also suppressed the release of Smac from the mitochondria in our study. Although the interaction between Smac and survivin in the mitochondria is not proven, this result can be explained by the activation of Bcl-2. It is reported that Bcl-2 attenuated the release of Smac from the mitochondria.16) We also observed that doxorubicin-induced cytochrome C release from mitochondria is inhibited by TAT-survivin protein transduction. Besides its interaction with Smac as already reported, survivin may also bind to other upstream proteins of Bcl-2 and regulate the release of mitochondrial proteins including Smac and cytochrome C. Taken together, a mechanism of action of survivin protein for the prevention of Smac and cytochrome C release can be explained by its direct effect on CREB in keeping the level of Bcl-2 high or by regulating the specific protein kinases that activate CREB.
According to previous studies, the transcriptional activity of CREB requires phosphorylation of serine 133 which provides positive conditions for binding with CRE.17) Also, CREB must form a complex such as its binding with transducers of regulated CREB protein 2 (TORC2) to achieve its transcriptional activity. The increase of protein kinase A (PKA) and protein kinase G (PKG)-Iα activity induces the translocation of TORC2 into the nucleus and also phosphorylates CREB.18) However, these intracellular protein kinases did not result in any change in the treatment of TAT-survivin (data not shown). We observed in this study that TAT-survivin transduction significantly attenuated the phosphorylation of p38 MAP kinase with doxorubicin treatment. We hypothesized that TAT-survivin could recover the CREB activation and Bcl-2 expression by suppressing p38, although our data did not provide direct evidence for a role of p38 in this event. MAP kinases are known to regulate gene expression, and cell mitosis, differentiation, survival, and apoptosis in response to a wide variety of stimuli.19) Generally, activation of extracellular signal-regulated kinases is involved in cardiac cell proliferation and survival.20) On the other hand, Jun N-terminal kinases (JNKs) are activated by several stimuli that induce cardiac myocyte death.21) Recent evidence has implicated the dual role of p38 as a regulator of cell growth, and thus it can promote either cellular survival or death.2) Many reports have suggested that p38 activation is necessary for cell death in response to various stimuli.22)23) In the opposite case, other observations have indicated that p38 plays a role in cell protection against several stresses.24)25) We observed that there was a significant increase in the phosphorylation of JNK in doxorubicin-treated cardiac cells; however, no detectable difference was observed in JNK phosphorylation upon TAT-survivin treatment (data not shown).
Several studies have shown that p38 MAP kinase can act a negative regulator of cardiac cell death against a number of stimuli.2)28)29) For example, p38 activation is associated with protection against cardiac cell death in a myocardial infarction model by coronary artery ligation of rats.26) Similarly, p38 activation protects cardiac cells from thermal stress-induced cell death in an isolated perfused heart model.27) Moreover, the p38 MAP kinase pathway is substantially involved in the protective effect against doxorubicin-induced cardiac cell death.2) Our results agree with these studies in that doxorubicin-induced p38 phosphorylation was inhibited by TAT-survivin transduction, leading to protection from cardiac cell death. These results were accompanied by the nuclear translocation of CREB sequestered in the cytoplasm, which results in the restoration of Bcl-2 expression. A previous study has also reported that activation of p38 MAP kinase in apoptotic cells correlates with reduced expression of Bcl-2 and survivin.28)
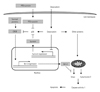
Thus, we propose that a possible mechanism underlying the protective effect of TAT-survivin is that doxorubicin-induced p38 activation may suppress other intracellular factors which activate the nuclear translocation of CREB and transcriptional activity of Bcl-2. A study has revealed that p38 MAP kinase is shown to crosstalk in the PKA pathway which is an upstream positive regulator of CREB.29) Our results support the hypothesis that doxorubicin-induced p38 activation can suppress the PKA pathway, leading to the inactivation of CREB and downregulation of Bcl-2 in H9c2 cardiac myocytes, and this event may be prevented by TAT-survivin protein transduction. Evidence has been reported that p38 MAP kinase mediates apoptotic cell death through downregulation of the Bcl family.30) The schematic diagram represents a model of the intracellular mechanism of the protective effect of survivin in doxorubicin-induced cell death (Fig. 5). Promising further studies may prove our hypothesis and reveal the biological role of survivin in cardiac cells.
In conclusion, our findings suggest the direct transduction of survivin protein into cardiac myocytes using the PTD system can prevent cell death due to doxorubicin. We can assume that the protective role of survivin protein is closely associated with the regulation of CREB activation and Bcl-2 expression, although its mechanisms are not fully elucidated.




 XML Download
XML Download