

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction; Risk of Familial Hypercholesterolemia
Familial hypercholesterolemia (FH), which occurs in one in five hundred (0.2%) Caucasians, is developed by low density lipoprotein receptor (LDLR) gene variations. It is a relatively common genetic disease that induces a rise in low density lipoprotein-cholesterol (LDL-C) and total cholesterol levels. However, this disease may easily be overlooked clinically.1)
According to the World Health Organization 1999 report, only fifteen percent of Canadians with this disorder were properly diagnosed. Thirty percent of these patients expired at the time of the first attack of myocardial infarction.2) Owing to the autosomal dominant inheritance of this disease, half of their offspring are also affected. Elevated serum cholesterol concentration which characterizes heterozygous FH leads to a greater than 50% risk of developing coronary heart disease in men by the age of 50 years and at least 30% risk in women by the age of 60 years. Furthermore, the fact that there is a high possibility of developing cardiovascular disease at young age calls for active clinical concern and vigorous effort.3)
The Characteristics of Familial Hypercholesterolemia
The pathophysiology of familial hypercholesterolemia
Familial hypercholesterolemia is an autosomal dominant genetic disease, and the clinical manifestation can result from mutation in either the LDLR gene, the APOB gene, or the PCSK9 gene. Among these, the most common genetic abnormality is the mutation in the LDLR located on chromosome 19.4)
Until now, more than one thousand mutations of the LDLR genes have been reported. Based on the expression levels and the activities of LDLRs, cases of FH homozygotes can be divided into two groups. One is a 'receptor-negative' group with less than 2% activity of LDLRs and another is a 'receptor-defective' group with 2-25% activity of LDLRs compared to that of unaffected subjects. In general, an inverse relationship is observed between LDLR activity and serum LDL-C level. In other words, lower activity of LDLRs implies less effective clearance of LDL particles. As a result, homozygous FH patients commonly demonstrate excessively high serum cholesterol levels, as much as 3-6 times those of normal individuals (total cholesterol level is unusually 500 mg/dL or higher). On the other hand, cases with heterozygotes inherited from only one parent would have 2-3 higher cholesterol levels depending on the extent of genetic abnormality and the effect of environmental factors (total cholesterol level is usually 300 mg/dL or higher).5)
Familial hypercholesterolemia and cardiovascular disease
Homozygous FH patients manifest cardiovascular symptoms before they reach the age of ten. Their symptoms are atypical, and sudden death is common. Receptor-negative patients may not survive over the age of twenty without proper treatment, while receptor-defective patients are inflicted with clinically significant cardiovascular disease before the age of thirty. Their serum cholesterol levels and clinical courses are mostly determined by their genetic background.6) The extent of mutation of LDLR genes is also a major factor that determines the natural course of heterozygous FH patients. However, uncontrolled risk factors for cardiovascular diseases such as smoking, hypertension and diabetes mellitus increase the risk considerably.7) It was reported that the attack rate of myocardial infarction in unidentified and untreated FH males and females before the age of sixty was sixty percent and thirty percent or greater, respectively.
Clinical features and diagnosis of familial hypercholesterolemia
Homozygous FH patients occur very rarely, happening in one in one million people. Most of these patients develop xanthoma before the age of ten. Unless proper treatment is undertaken as mentioned earlier, most patients expire from cardiovascular disease before reaching adolescence. Thus, in reality, it is not easy to change the clinical course. Heterozygous FH patients occur in one in five-hundred people. Therefore, it requires particular clinical attention. Accurate statistics are not yet available in the Korean population. An assumption was made that the occurrence rate may be less in Korea than that of other countries. A total cholesterol level of 300 mg/dL or higher is typical in reported cases. Clinicians should be suspicious of FH in the presence of severe hyperlipidemia, and should verify the family history of hyperlipidemia and cardiovascular disease. It was reported that the diagnostic specificity can be up to 98% using only family history and cholesterol level. For instance, a male patient with a total cholesterol level of 290 mg/dL or higher is enough to be suspicious of familial hyperlipidemia (Table 1). Moreover, any patient with a cholesterol level of 240 mg/dL or higher in the presence of familial hyperlipidemia or past medical history of cardiovascular disease would have a high diagnostic possibility of FH. In such case, causes of secondary hyperlipidemia, such as hypothyroidism or nephritic syndrome, should be preferentially excluded.8)
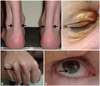
Tendon xanthoma occurs in three fourth of middle-aged patients and is largely discovered in the Achilles tendon. It may also occur in the extensor area of the dorsum of the hand, heel or the knee. Since xanthoma may be manifested only to the extent of thickening of a tendon, FH patients may be overlooked by clinicians without due concern. Xanthelasma or early arcus cornealis may also be observed (Fig. 1).9)
Several diagnostic criteria for heterozygous FH have been suggested. One of them10) is shown in Table 2. Familial hypercholesterolemia should be considered in adults with a total cholesterol level of 285 mg/dL or higher and clinically diagnosed based on the extent of hyperlipidemia and family history of early cardiovascular disease.11) On the other hand, a patient with a past medical history of early cardiovascular disease should undergo a screening test for his or her family members. The criteria have a high specificity of 90% for 'definitive' diagnosis, but have a low sensitivity of only 35%. On the contrary, the sensitivity for 'possible' diagnosis was high at 90%, while the specificity was low at 30%.12) Definitive diagnosis, which is possible by testing mutations of the related genes, is only available in certain laboratories. In addition, costs for definitive tests are not undemanding. Thus, clinical concern and suspicion are important in making the diagnosis of FH.
Current treatment of familial hypercholesterolemia
Several significant clinical trials have been carried out for patients with FH.13) First, there is ASAP, "Atorvastatin vs. Simvastatin on Atherosclerosis Progression", as an interventional study using statin. The ASAP study, which included 325 FH patients, compared a high dose regimen of atorvastatin 80 mg with the conventional dose regimen of simvastatin 40 mg.
The carotid intima media thickness (CIMT) was increased in the simvastatin-administered group. On the other hand, the thickness statistically significantly decreased in the atorvastatin-administered group. The group administered with a high-dose of atorvastatin showed a fifty percent decrease in LDL-C levels, while the group with simvastatin administration revealed a forty percent reduction.
The Exetimibe and Simvastatin in Hypercholesterolemia enhances atherosclerosis regression (ENHANCE) study in 2008 was a study including 720 FH patients. It investigated the effect of a single simvastatin regimen and that of a combination therapy of simvastatin and ezetimibe. The results showed reduced rates in LDL-C levels for the single regimen group with simvastatin 80 mg and the group with a combination therapy of simvastatin 80 mg and ezetimibe 10 mg were 41% and 58%, respectively. The group with the combination therapy showed further reduction of twenty-six percent in high sensitivity-C-reactive protein. However, different from what was anticipated, there were no differences in CIMT between these two groups. As the ENHANCE study reported, it triggered many debates regarding the utility of CMIT or the anti-atherosclerotic effect with respect to drug interventional studies for FH patients.
However, a few precautions should be taken when interpreting a comparative analysis of a failed ENHANCE study and a successful ASAP study. In the ENHANCE study, about eighty percent of subjects were administered statin before they were enrolled in the study. Unexpectedly, the mean CIMT of these patients was 0.69 mm. This value is within the normal level. This could be explained by the effect of existing statin treatment. Administration of statin prior to the study may already stabilize CIMT, and the carotid intima media may take a normal form. In such cases, additional drug treatment would not revert its progress. The mean CIMT of ASAP study subjects, in whom the CIMT decrease was verified, was 0.925 mm. In fact, a thicker CIMT before the study predicts more distinctive reduction of CIMT after drug intervention.
The ENHANCE study, in which changes of a surrogate marker, CIMT, were observed, could not provide a clear-cut answer to the additive effect of ezetimibe. The recently reported positive results of the Study of Heart and Renal Protection shed light on the future of ezetimibe. However, depending on the results from IMProved Reduction of Outcomes: Vytorin Efficacy International Trial, which will be reported in 2013, it will enter upon a new phase and definitively change the situation. Until then, the principle of treatment for FH be suggested as follows.
In cases where the target LDL-C level is difficult to achieve or a high-dose statin treatment cannot be continued due to adverse reactions, a combination therapy with concurrent ezetimibe administration must be considered.
Efficacy of ezetimibe in the treatment of familial hypercholesterolemia
Unlike general hypercholesterolemia, FH is not easy to treat with dietary management or statin therapy alone.14) Moreover, it is difficult to expect the increasing effect of its manifestation by statin.
In such circumstances, a more effective LDL-C lowering effect may be attained by concurrent treatment with ezetimibe that has a distinct pharmacologic mechanism, through which cholesterol absorption is suppressed in the intestine. However, such drug treatment is not sufficient for homozygous patients. Only about a thirty percent drop in LDL-C level would be attained even with a combination therapy of high-dose statin and ezetimibe. Thus, LDL apheresis or liver transplantation can also be attempted.
Heterozygous hyperlipidemia patients may reduce their cholesterol levels by 10-15% through dietary adjustment. Although rare, a high dose statin regimen may achieve the target level. However, dose reduction may be required due to adverse reactions such as myalgia. Therefore, a combined therapy with excellent cholesterol-lowering effect and less adverse reactions should be considered. By the same token, ezetimibe may be a first-rate adjunct pharmacologic agent. In reality, concurrent ezetimibe treatment along with statin therapy in patients with FH may additionally reduce LDL-C levels by 10-20% (Table 3).
Additional genetic background in familial hypercholesterolemia; Lipid change by mutations of PCSK9 genes
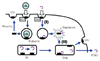
PCSK9 genes (Fig. 2) are largely involved in recycling of LDLRs. PCSK9 is a serine protease synthesized in the liver and intestines. Well-functioning PCSK9 proteins inhibit recycling of LDLRs, and LDL and other lipid substances, that are mediated through LDLRs, may not be removed smoothly, finally causing higher LDL-C levels. Any mutations that cause gain-of-function of PCSK9 may result in LDL-C level as shown in FH cases. According to recent studies by Brown and Goldstein et al., loss-of-function mutations of PCSK9 gene were mostly discovered among the black population, and their mean LDL-C levels were found to be lower by 10-20 mg/dL.15) Although the differences in the values were not big, the amount of lifetime exposure may affect them immensely. Accordingly, a seventy percent decrease in the development of cardiovascular disease was reported in such families.
Recently, neutralizing monoclonal antibody against PCSK9 protein has been developed and reportedly reduces LDL-C level by 30-40% in FH cases.
Conclusion Remarks
In spite of substantial limitations in the diagnosis and management of lipid profiles in FH patients, it is important to identify and manage the condition of FH in order to prevent atherosclerotic cardiovascular disease. With the help of progress in drug development, novel therapeutic strategies will be available soon and more ideal LDL-C levels can be achieved and maintained with minimal additional efforts. Functional inhibition of the PCK9 protein and suppression of apoB100 production are the examples.
The updated European Society of Cardiology guideline in 2011 states that the severe form of dyslipidemia or familial dyslipidemia should be considered as high risk for cardiovascular disease, suggesting that physicians should pay more attention in the diagnosis and management of dyslipidemia patients with family history including FH.



 XML Download
XML Download