

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Hereditary hemorrhagic telangiectasia (HHT), also known as Rendu-Osler-Weber disease, is an autosomal dominant disorder characterized by atriovenous malformations (AVMs) and telangiectases in multiple organs. The reported incidence of this disease is 1/10000 in North America but it varies between studies.1) The pathologic mechanism includes primary dilatation of postcapillary venules resulting in a direct connection between the arterial and venous systems, bypassing capillaries and finally forming arteriovenous shunts.2) The most common clinical features are nosebleeds and telangiectases on the lip, oral mucosa and hands.
High-output cardiac failure is a rare complication of HHT3) usually caused by shunting of blood through AVMs in the liver. We describe two cases of high output heart failure due to large hepatic AVMs, highlighting their importance in the differential diagnosis of heart failure syndromes.
Case
Case 1
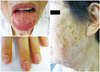
A 68-year-old woman came to the hospital because of dyspnea {New York Heart Association (NYHA) class 3}. On physical examination, the patient was puffy and anemic. Multiple telangiectases were found on the lips, tongue and finger tips (Fig. 1). Her blood pressure was 100/70 mm Hg, she had a heart rate of 94/min, and a respiratory rate of 22/min. Cardiac examination revealed a midsystolic murmur along the sternal border and a bruit was audible over the epigastrium. Her medical history was positive for recurrent epistaxis and multiple episodes of gastrointestinal bleeding. She stated that her family members (her mother, one sister and one daughter) also had suffered from recurrent epistaxis.
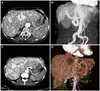
The laboratory work-up revealed hypochromic microcytic anemia (hemoglobin 8.8 g/dL), normal liver function tests, and normal prothrombin time. Chest X-ray showed generalized cardiomegaly (CT ratio 65%) with increased pulmonary vascular markings and pulmonary edema (Fig. 2). Transthoracic echocardiogram showed a dilated hypercontractile left ventricle and a restrictive mitral inflow pattern (mitral E to A ratio=1.3, E/e=16.6). CT angiography of the abdomen showed a tortuous and dilated hepatic artery and near total replacement of liver parenchyma by engorged vascular structures with arteriovenous shunt formation (Fig. 3A and B). Cardiac output by right heart catheterization was 12.47 L/min, capillary wedge pressure was 22 mm Hg and pulmonary artery pressure was 45/18 mm Hg. MRI of the brain with contrast showed an aneurysm (7 mm) in the right paraclinoid intracranial artery (ICA) and was managed with Guglielmi detachable coils (MicroVention, Aliso Viejo, CA, USA). After correction of anemia by iron supplements and treatment with angiotensin receptor blocker and diuretics, heart failure symptoms were significantly improved.
Case 2
A 56-year-old woman was referred for dyspnea (NYHA class 3). She had been diagnosed with liver cirrhosis and hypertensive heart failure 8 years previously and was intermittently managed with angiotensin receptor blocker and diuretics. From a young age she had experienced frequent epistaxis. She also had multiple episodes of gastrointestinal bleeding since the age of 49 years. The last bleeding episode occurred 1 month ago. Gastrointestinal endoscopy showed multiple telangiectases in the gastric mucosal walls which were electrocoagulated. Her family history revealed that her father and brother also experienced recurrent epistaxis. Her father died at the age of 60 years due to cerebral hemorrhage. Her blood pressure was 130/60 mm Hg, she had a heart rate of 96/min, and a respiratory rate of 20/min. On physical examination, several telangiectases were found in both cheeks, and bounding pulses were palpated. The liver tip was palpable below the right costal margin. A systolic murmur along the sternal border and a loud bruit over the whole upper abdomen were heard.
Her laboratory work-up revealed hypochromic microcytic anema (hemoglobin 10.6 g/dL), normal liver function tests, and normal prothrombin time. Hepatitis viral markers were negative. Chest X-ray showed generalized cardiomegaly (CT ratio 65%) and pulmonary edema. Transthoracic echocardiogram showed a dilated and hypertrophied left ventricle with hypercontractile left ventricular systolic function with signs of increased filling pressure and mitral inflow pattern (E/A ratio=1.5, Deceleration time of mitral inflow E=157 msec, E/E'=12.8). In order to identify the possible arteriovenous connection inside the abdomen, abdominal CT angiography was performed. This showed a markedly engorged hepatic artery and vein with a fistulous connection between the two (Fig. 3C and D). Based on this finding, the diagnosis of high output heart failure caused by extensive hepatic arteriovenous connections was made. For identifying the accompanying vascular abnormalities, chest CT angiography and cerebral MR angiography were performed and chest CT angiography showed a small AVM in the right middle lobe of the lung (Fig. 4). Currently, her condition has been stabilized; however, the possibility of liver transplantation in case heart failure becomes intractable has been explained to the patient.
Discussion
Hereditary hemorrhagic telangiectasia or Osler-Weber-Rendu disease, characterized by telangiectases and AVM in multiple organs is a rare disease with autosomal dominant transmission.4) The classic triad is recurrent epistaxis, mucocutaneous telangiectasia, and familial occurrence. The diagnosis of HHT is confirmed if any three of the following four criteria are met: 1) recurrent epistaxis; 2) multiple telangiectasias; 3) presence of visceral lesions (in the gastrointestinal tract, lung, liver, brain); 4) first degree family history of HHT.4)
Hepatic involvement in HHT has been reported in 74% of patients with CT of the abdomen. But liver involvement tends to be asymptomatic in majority of cases and the frequency of symptomatic hepatic involvement is less than 10%.1) Although heart failure secondary to the hyperdynamic circulatory situation caused by arteriovenous fistulas is a well-known clinical entity, the clinical presentation of heart failure is very rare in HHT.3) The physiological mechanism in high output heart failure is of reduced systemic vascular resistance. This leads to activation of the renin-angiotensin-aldosterone system, causing salt and water retention. The shunting of blood through the AVMs, especially in the liver leads to maldistribution of cardiac output. In order to supply blood to vital organs, cardiac output is increased with elevated stroke volume and heart rate, leading to high output heart failure. A high cardiac output has been described as being >8 L/min or a cardiac index >3.9 L/min/m2.5) Right heart catheterization in our case 1 demonstrated a markedly increased cardiac output of 12.47 L/min. Reported cases of right-heart catheterization in patients with symptomatic hepatic AVMs have calculated shunts of 24% to 58% of the cardiac output.6) The echocardiography usually shows compensatory left ventricular dilatation and left ventricular hypertrophy, but preserved or high left ventricular systolic function. 5) High output heart failure could be presented as a diastolic dysfunction.7) Echocardiographic findings in our cases were consistent with those of the previous reports.
In contrast to low output heart failure, clinical trial data regarding treatment of high output heart failure are lacking. The treatment of high output heart failure in patients with HHT is usually conservative. Diuretics are the mainstay of treatment in high output heart failure, especially with symptoms of volume overload. The patients in the two above mentioned cases were treated with angiotensin receptor blocker and diuretics. The treatment was effective for managing their symptoms related to heart failure. This treatment may not be effective in patients with large vascular malformations. There have been scattered experiences with transfemoral embolization,8) surgical ligation of the feeding vessels9) in symptomatic patients. However, in intractable patients, liver transplantation10) has been the standard treatment. Recently, treatment with bevacizumab11)-the anti-vascular endothelial growth factor has been reported to be effective in heart failure patients with HHT by slowing the progression of vascular malformations.
Cerebral AVMs are present in 5% to 23% of patients with HHT with intracranial hemorrhage rates of 0.41% to 2% per year.1) But the association between intracerebral aneurysm and HHT has not been reported in the literature. Thus, the possibility of an incidental coexistence of intracerebral aneurysm with HHT is likely. Reported cases with cerebral involvement were managed by neurovascular surgery, embolotherapy, and stereotactic radiosurgery.12)13) The case of the woman with a cerebral aneurysm in the right paraclinoid ICA was treated successfully with Guglielmi detachable coils.
Recently, findings from genetic studies have been reported. HHT is caused by a disturbance of the transforming growth factor signaling pathway. Three HHT genes (Endoglin, Activin A receptor type II-like 1, SMAD4) have been identified to date, and several others are suspected.12) Genetic studies were not performed in our cases due to economic problems.
The overall prevalence of HHT in North America is estimated at 1 : 10000. However, the prevalence may be underestimated because many cases may be asymptomatic. When a proband is found, it is necessary to take a careful family history in order to detect possible HHT in multiple relatives. Although the disease may be easily suspected when history and clinical manifestations are typical, there are some cases like those in our study that were initially misdiagnosed. The most important first step towards managing HHT is clinical suspicion of HHT and early diagnosis by detailed history taking and physical examination, because the first presentation of this disease can be a critical condition such as massive hemoptysis, brain hemorrhage.
In conclusion, we present here two patients of HHT who presented with high output heart failure. High output heart failure was controlled by medical management with some clinical improvement, but a careful follow-up may be necessary to detect developing cerebral or pulmonary AVMs. Clinical suspicion of HHT based on detailed history taking and physical examination is essential for early detection and proper management of heart failure associated with HHT.


 XML Download
XML Download