

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Deep vein thrombosis (DVT) is an important cause of morbidity and mortality with an estimated annual incidence of 0.67 to 1.0 per 1000 among the general population in Western countries.1-3) Pulmonary embolism and post-phlebitic syndrome are the recognized complications. DVT may be caused by hereditary or acquired defects in the coagulation-anticoagulation pathway. Risk factors include prolonged non-ambulation, recent surgery, obesity, prior DVT, female sex hormone therapy, post-partum status, lower limb trauma, neoplastic disease and stroke. Recurrent DVT, a positive family history and unusual presentation may warrant investigation for hereditary defects.4)
Case
A 40-year-old non-smoker, non-diabetic, normotensive male presented with swelling and aching pain of the left lower limb extending from the left thigh to the ankle over one week. Upon examination, his pulse was 80 beats per minute and his blood pressure was 140/85 mm Hg. The left lower limb with dusky blue discoloration of skin was diffusely swollen, warm, and exquisitely tender to touch. Arterial pulses in the legs were palpable. Three years ago, he developed a similar illness in the right leg, and was diagnosed as a case of DVT (Fig. 1).
Initial heparin therapy was followed by warfarin, which was discontinued after three months. Within two years the patient had DVT in the left lower limb (Figs. 2 and 3). The patient was prescribed an oral anticoagulant that was discontinued after several months.
There was no ulceration or gangrenous changes, nor was there any lymphadenopathy. Examination of the precordium revealed normal findings. Other systems were also normal. There was no neurological deficit. Fundoscopy was normal.
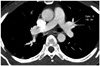
A duplex study of the lower limb vessels showed acute DVT involving the left femoro-popliteal segment. CT angiography revealed normal pulmonary arteries (Fig. 4). Blood sugar, renal and liver function tests, serum electrolytes and calcium were normal. A haemogram including platelet count, urinalysis, chest radiograph and echocardiography revealed normal findings. A compliment fixation test for filaria was negative. Because of the recurrence of venous thromboembolism (VTE), it was decided to search for any coagulation abnormality. Therefore, the blood coagulation profile was studied four weeks after the onset of DVT. Platelet count, bleeding time, clotting time and prothrombin time (PT) were normal. In addition, plasma fibrinogen and homocysteine levels were within normal range, and antithrombin activity in the blood was also normal. Conversely, the protein C activity was 58% (normal range: 70-140%), and the protein S activity was greatly reduced to 17% (normal range: 54-137%). Ideally, coagulation assay should be conducted one month after completion of a course of anticoagulation, but considering the number and frequency of recurrence of DVT, and associated risks of life-threatening complications like pulmonary thromboembolism, anticoagulant therapy was not stopped.
In the acute setting, the patient was managed according to standard protocols, including elevation of the affected limb. Additionally, 60 mg of enoxapain was administered subcutaneously every 12 hours for one week, and this was overlapped with 5 mg warfarin everyday 72 hours after the onset of low-molecular-weight heparin therapy. The international normalized ratio (INR) was kept between 2 and 3. The patient gradually improved and was discharged with warfarin. In a subsequent visit one month later, the INR was found to be 2.8; therefore, the patient was advised to continue oral anticoagulant therapy indefinitely.
Factor V Leiden and mutant prothrombin (Prothrombin 20210A) assay could not be done because these tests are not available in Bangladesh.
During follow-up over the next two-years, the patient was doing well and did not have any complaints related to DVT.
Discussion
Thrombophilia is the term used to describe predisposition to an increased risk of venous and occasionally arterial thromboembolism owing to haematological abnormalities.5) This condition may be acquired or hereditary. A genetic predisposition to thrombophilia has been identified in up to one-third of unselected patients with VTE and more than one-half of cases with familial thrombosis.6)7) There may also be an inability of the body to produce adequate amounts of normal protein, or the body may produce abnormal protein that does not function normally. Multiple defects can often be found in patients with the most marked thrombotic predispositions.6)8) The presence of factor V Leiden, prothrombin gene mutation, antithrombin III, protein C or protein S deficiency, hyperhomocysteinemia, hyperfibrinogenemia and elevated lipoprotein (a) are important causes of inherited thrombophilia.
Deficiencies of the endogenous anticoagulants, protein C, protein S and antithrombin III, are usually polygenic and have an autosomal dominant inheritance with variable penetrance.9) The aggregate prevalence of the three conditions is approximately 7% in DVT patients.10)11) Their deficiency increases the risk of venous thromboembolism. Protein C is made in the liver and is vitamin K dependent. It acts to inactivate factor V and factor VIII. This protein also requires protein S as a cofactor and is activated by thrombin.12) Protein C deficiency is observed in 1 in 200 to 1 in 300 healthy subjects, many of whom remain asymptomatic.13) The protein S is vitamin K dependent and is synthesized by hepatocytes and megakaryocytes. Its deficiency has a prevalence of 1 to 2% in patients with DVT.10)11) Antithrombin, which is made in the liver and endothelial cells, inactivates thrombin and other serine proteases. The prevalence of antithrombin III deficiency in the general population is 0.02%, and in patients with a history of VTE is upto 4%.14) In the case presented here, both protein C and protein S were deficient, which presumably made the patient more likely to develop recurrent DVT. Hao et al.15) reported a case of a 44-year-old Japanese male who had three events of DVT within four years and was later found to have antithrombin deficiency.
The prevalence of factor V Leiden is 2-15% in the general population,14) but it is rare in native Africans and Asians.16) DVT is the most common clinical manifestation of factor V Leiden. Prothrombin G20210A is seen in 1-4% of general population,14) and patients with the 20210A allele have significantly higher levels of plasma prothrombin, which is believed to mediate the procoagulant effect.9)
Hyperhomocysteinemia may be caused by abnormal homocysteine metabolism due to genetic or nutritional defects.17) There is increased risk of both venous and arterial thrombosis.9)
The first step in the diagnostic approach to all patients with venous thrombosis consists of a careful review of their history and physical examination combined with routine laboratory testing to characterize the severity of the thrombotic condition and determine the presence of any of the acquired causes of hypercoagulability. The second step is to consider screening for the causes of hereditary and acquired thrombophilia in selected patients.18) Indications for thrombophilia screening are 1) all patients with a first episode of spontaneous VTE, 2) patients with VTE under the age of 50 years, even when they have a transient predisposing factor, 3) patients with VTE whose only risk factor is oral contraceptive therapy, estrogen replacement therapy or pregnancy, 4) patients with recurrent VTE, irrespective of risk factors, 5) patients with recurrent superficial thrombophlebitis without cancer and in the absence of varicose veins, 6) patients with VTE at unusual sites such as the cerebral venous sinus, mesenteric or hepatic veins and retinal vein occlusion under the age of 50 years, 7) patients with warfarin-induced skin necrosis or neonates with purpura fulminans not related to sepsis, 8) asymptomatic first degree relatives of individuals with proven symptomatic thrombophilia, 9) two consecutive or three non-consecutive abortions at any gestational age or one fetal death after the 20th week, 10) severe pre-eclampsia, 11) children with VTE.5)
Functional, immunological and molecular tests are available for laboratory evaluation of hereditary thrombophilia. Functional and immunological investigations are used for antithrombin, protein S or protein C deficiency and factor V Leiden. Their sensitivity and specificity are variable and are influenced by pregnancy, hepatic insufficiency and medications. Molecular tests are gene specific and more sensitive and specific and available for factor V Leiden and mutant prothrombin 20210A.5) A full blood count, platelet count, PT, activated partial thromboplastin time (aPPT) and thrombin clotting time (TCT) should be conducted routinely. Liver function tests and blood urea and electrolytes are also indicated.14) Blood counts may indicate myeloproliferative disorders, while APTT may be helpful in antiphospholipid antibody syndrome, PT for interpretation of low protein C or protein S, and TCT for dysfibrinogenemia. Antiphospholipid antibodies should also be included. Further investigations under appropriate circumstances include assays for antithrombin, protein C and protein S, modified APC/SR test for factor V Leiden and polymerase chain reaction (PCR)-based assay for prothrombin G20210A. Factor VIII, homocysteine and fibrinogen assays are the second-line tests for thrombophilia.14) Polymerase chain reaction for factor V Leiden or prothrombin 20210A can be conducted at any time.5) Conversely, antithrombin, protein C and protein S assays are best delayed until at least one month after completion of anticoagulation.14)
Heparin, either unfractionated or low-molecular-weight, and warfarin are the mainstays of treatment of acute DVT. The duration of secondary prophylaxis with warfarin depends on the presence of risk factors associated with recurrent DVT. Thrombophilia due to deficiency of antithrombin, protein C or protein S, homozygosity for factor V Leiden or prothrombin G20210A, presence of cardiolipin antibodies or lupus anticoagulant, elevated factor VIII or homocysteine and combined defects requires prophylaxis with warfarin indefinitely. INR is kept between 2 and 3.19) In a patient with recurrent DVT with thrombophilia, secondary prophylaxis with warfarin is of the utmost importance. The repeated episodes of DVT observed in the case presented here could probably have been prevented if warfarin were continued indefinitely after the first recurrence.
In conclusion, in patients with recurrence or unusual clinical presentation of DVT, hereditary coagulation defects should be suspected. Laboratory tests during the acute episode may produce considerable confusion; therefore, they should be conducted at least one month after the cessation of anticoagulant therapy. In the presence of significant hereditary thrombophilia, secondary prophylaxis with warfarin is lifelong in most cases.


 XML Download
XML Download