

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Platelets play a key and beneficial role for primary hemostasis on the disruption of the integrity of vessel wall. Platelets adhere to the exposed subendothelial collagen through the interaction of circulating von Willebrand factor and glycoprotein (GP) Ib-IX-V complex, a major platelet membrane receptor responsible for adhesion.
After the adhesion of platelets to the subendothelial collagen, platelets become activated. Activation processes of platelets include the release of adenosine diphosphate (ADP) from dense granules, the synthesis of thromboxane A2 (TXA2) via a series of enzymatic reactions, and thrombin formation via a coagulation cascade. In addition, ADP is released from endothelial cells at the site of vascular injury. Binding of these mediators to G protein-coupled receptors of platelet membrane, changes the shapes of platelets from smooth discs to irregular spheroids with extrusion of filopodia, and activates platelet membrane GP IIb/IIIa receptors through complex signal transduction mechanisms. Activated GP IIb/IIIa binds to fibrinogen, finally forming platelet aggregates.1)2)
Platelet aggregation is not only an essential part of hemostasis, but also initiates acute coronary syndrome or ischemic stroke. Recent efforts to prevent the development of unstable angina or acute myocardial infarction from stable atheromatous plaques, and thrombotic complications related to percutaneous coronary intervention progress the understanding of mechanisms of platelet activation, but its precise mechanism and related proteins are not fully identified. This review will focus on the recent progress of knowledge about platelet activation mechanism.
Mediators of Platelet Activation
Adenosine diphosphate
Adenosine diphosphate and its analogues bind to P2 receptors, which are expressed ubiquitously throughout the human body including on platelets, on vascular smooth muscle cells, and on endothelial cells. P2 receptors are divided into P2X and P2Y receptors. P2X receptors are ligand-gated ion channel receptors, and P2Y receptors are G-protein coupled receptors. Each subtype of P2X and P2Y receptors is classified by the relative potency of purine and pyrimidine nucleotides, as agonists, to induce intracellular calcium transients or smooth muscle contraction after genetic cloning. Seven P2X receptors (P2X1, P2X2, P2X3, P2X4, P2X5, P2X6, and P2X7) and P2Y receptors (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, and P2Y13) are cloned in mammalian cells.3) Platelets express P2X1, P2Y1, and P2Y12 receptors.4-6)
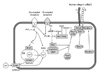
Adenosine diphosphate binding to P2X1 receptor results in rapid extracellular calcium influx, leading to alteration of platelet shape, but without platelet activation. ADP-stimulation of Gq-coupled P2Y1 receptor activates phospholipase C (PLC) and induces a transient increase in intracellular calcium concentration resulting in platelet shape change and in weak, transient platelet aggregation. Stable and firm platelet aggregation requires ADP binding to P2Y12 receptor and activation of GPIIb/IIIa receptors in the platelets membrane.7) Activation of Gi-coupled P2Y12 receptor liberates the Gi protein subunits, αGi and βγ. The subunit αGi decreases the platelet cyclic adenosine monophosphate (cAMP) level through the inhibition of adenylyl cyclase. This decrease in cAMP production leads, in turn, to a reduction in the activation of protein kinase C (PKC), and, after complex signal transduction processes, activation of GP IIb/IIIa receptors (Fig. 1). The subunit βγ activates the phosphatidylinositol 3-kinase (PI-3K), which regulates protein kinase Akt and contributes to the activation of GP IIb/IIIa receptors.
The P2Y12 receptor is inhibited by ticlopidine, clopidogrel, prasugrel, or ticagrelor, which are currently used for patients with acute coronary syndrome or coronary stents to prevent thrombotic complications.
Thrombin
Thrombin is the most potent activator of platelets. Thrombin formation is initiated by the exposure of tissue factor to the coagulation system after disruption of the vascular endothelium. Thrombin formation takes place on cellular surfaces, including the phospholipid membrane of activated platelets, which amplifies coagulation processes. Thrombin interacts with protease-activated receptors (PARs) on the surface of human platelets, which couple to Gq, G12/G13 and Gi family of heterotrimeric G proteins.8) Of the 4 PARs, human platelets express PAR1 and PAR4. PAR1 mediates human platelet activation at low thrombin concentrations, whereas PAR4 induces platelet activation only at high thrombin concentrations.9)
Thrombin binding of PARs initiates the cleavage of the extracellular domain of the receptor and exposure of a tethered ligand at the new end of the receptor. Signaling through either PAR uses PLC pathway and PKC pathway, leading to the activation of GP IIb/IIIa receptors.10) In addition, interaction of thrombin with PARs causes the production of TXA2, the release of ADP, and generation of more thrombin on the platelet surface.
Vorapaxar is an oral reversible PAR1 antagonist. In a phase 2 trial, vorapaxar showed a trend toward lower incidence of major adverse cardiovascular events (MACE) without differences of major and minor bleeding in patients scheduled for angiography and possible coronary stenting.11) However, in a recent large randomized clinical trial, the addition of vorapaxar to standard therapy in patients with non-ST elevation acute coronary syndrome did not reduce MACE but significantly increased the risk of major bleeding, including intracranial hemorrhage.12)
Thromboxane A2
Thromboxane A2 is produced from arachidonic acid through conversion by cyclooxygenase-1 and thromboxane synthase. Like ADP, TXA2 functions as a positive-feedback mediator during platelet activation. Human TXA2/prostaglandin (TP) receptor exists in 2 isoforms, TPα and TPβ, and human platelets express both isoforms.13) TP receptor couples to Gq and G12/G13. TXA2 stimulation of Gq proteins causes activation of PLC, resulting in accumulation of inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). Stimulation of G12/G13 proteins activates Rho signaling. TP receptor-mediated platelet shape change is mainly dependent on G12/G13, while aggregation is dependent on Gq.14)
Platelet Integrins
Integrins are a widely expressed family of heterodimeric transmembrane receptors linking extracellular ligands to intracellular signaling pathways and contributing to a wide variety of physiological processes. They are composed of α- and β-subunits, which are non-covalently linked to each other. Both subunits consist of a large N-terminal extracellular domain forming a globular head, a single transmembrane domain and a short cytoplasmic tail domain. Platelets express 3 different β1-integrins: α2β1 (collagen receptor), α5β1 (fibronectin receptor), and α6β1 (laminin receptor); and 2 β3-integrins: αIIbβ3 and αvβ3. Although integrin α2β1 is associated with platelet adhesion to the injured vessel wall, the dominant integrin on the platelet surface is αIIbβ3, which mediates platelet aggregation.15)16)
Integrin αIIbβ3 is present 60,000-80,000 copies per resting platelet, which is the highest density of all platelet membrane proteins. The membranes of platelet α-granules contain αIIbβ3 that becomes externalized on platelet secretion to increase the surface expression of αIIbβ3 by 25% to 50%. The αIIb subunit consists of 1008 amino acids, which is composed of a heavy and a light chain. The light chain contains a 20-amino acid cytoplasmic tail, a transmembrane helix, and an extracellular segment that is a disulfide linked to the heavy chain, which is entirely extracellular. The β3 subunit is a single polypeptide chain of 762 amino acids. The 2 subunits assemble into the divalent, cation-dependent heterodimer during biosynthesis in megakaryocytes (Fig. 2).17)18)
The ligands of αIIbβ3 include fibrinogen, fibronectin, von Willebrand factor, and vitronectin. Fibrinogen and von Willebrand factor support platelet aggregation. Integrin αIIbβ3 recognizes Arg-Gly-Asp (RGD) sequence of the ligands. The most important characteristic of integrin αIIbβ3 is the affinity modulation after its activation. In resting platelets, the affinity of αIIbβ3 for fibrinogen, a main ligand of platelet aggregation, is low, and minimal binding occurs despite the high levels of fibrinogen in blood. Activation of integrin αIIbβ3 depends primarily on the conformational change of the receptors, whether induced by platelet adhesion to extracellular matrix, or agonists, such as ADP, thrombin, or arachidonic acid. This requires transmission of information from within the cell to the extracellular domain of αIIbβ3, a process referred to as "inside-out" signaling, which changes receptors from a low- to high-affinity state (Fig. 3A). Integrin activation can involve changes not only in affinity for ligand, but also in avidity for ligand, a consequence of receptor clustering. Both mechanisms can lead to activation of αIIbβ3, but affinity modulation is dominant. Platelet activation leads to a marked increase in the affinity of αIIbβ3 for fibrinogen. Ligand-occupied integrin αIIbβ3, on the other hand, triggers various cellular processes, such as reorganization of the cytoskeleton within platelets, through "outside-in" signaling (Fig. 3B).17)
Activation Mechanism of Integrin αIIbβ3
The activation mechanism of integrin αIIbβ3 from a low- to high-affinity state has been intensively studied because this signal transduction pathway may be a potential target for a new antithrombotic agent. Conformational changes of the extracellular domains of integrin αIIbβ3 and resulting affinity modulation are initiated by the interaction at the cytoplasmic tails. Cellular control of integrin activation requires transmission of a signal from the small cytoplasmic tails to the large extracellular domains. The exact mechanism involved in inside-out and outside-in signaling of integrin αIIbβ3 is not fully understood, and the functional significance of the related proteins is still evolving (Fig. 4).10)16)
Rap1b/CalDAG-GEFI
Recent studies have emphasized the role of the Rap family of small GTPases in inside-out signaling of integrin activation. Rap1b is the most abundant Ras family member in platelets.19) Rap1b deficiency in murine platelets showed reduced platelet aggregation, decreased activation of integrin αIIbβ3, and protection from arterial thrombosis.20) Rap proteins cycle between GDP-bound inactive form and GTP-bound active form. GTP binding to Rap is facilitated by guanine nucleotide exchange factors (GEFs). In platelets, Rap1b is controlled by Ca2+ and diacylglycerol-regulated guanine nucleotide exchange factor I (CalDAG-GEFI), which contains binding sites for Ca2+ and DAG, and a GEF domain.21) The importance of CalDAG-GEFI was demonstrated in CalDAG-GEFI-deficient mice. Cal-DAG-GEFI deficiency resulted in impaired platelet aggregation responses to ADP or TXA2, but not to collagen or thrombin.22) CalDAG-GEFI-/- mice showed prolonged bleeding time and protection against experimental thrombosis.
Different agonist responses in CalDAG-GEFI-deficient platelets suggest the existence of a CalDAG-GEFI-independent mechanism of integrin αIIbβ3 activation. In murine platelets, PAR4 agonist-induced aggregation in the absence of CalDAG-GEFI required cosignaling through the αGi-coupled P2Y12 receptor involving PKC. Therefore, PKC was identified as an alternative pathway leading to Rap1 and αIIbβ3 activation. It has been proposed that rapid, but reversible activation of Rap1 is mediated by CalDAG-GEFI, whereas PKC ensures sustained Rap1 activation through its central role in the release of ADP from dense granules.21)
Agonist activation of platelets is usually accompanied by an increase in the cytosolic Ca2+ concentration, which is required for integrin activation and releases of the second wave mediators, such as ADP or TXA2. The source of Ca2+ can be either intracellular or extracellular. Agonists binding of platelet Gq coupled-receptors activates PLC, which in turn produces inositol 1,4,5-triphosphate (IP3) from phosphoinositol biphosphate (PIP2). The intracellular Ca2+ is released from the sarcoplasmatic reticulum (SR) by IP3. Because of the limited Ca2+ store of the ER, the major source of an intracellular Ca2+ transient is store-operated Ca2+ entry (SOCE) of the extracellular Ca2+ through the activation of Ca2+ release activated calcium channels (CRAC), Orai1, in the platelets membrane, which are controlled by the depletion of SR Ca2+ stores.23) Recent studies identified stromal interaction molecule 1 (STIM1) as a key regulator of SOCE into platelets.24) STIM1 is a type I transmembrane protein containing a calcium binding EF hand motif, which is occupied by Ca2+ in resting platelets. After platelet activation, Ca2+ is released from the ER and STIM1 is no longer occupied by Ca2+, resulting in translocation of STIM1 within the platelets to allow SOCE by opening the Orai1. Platelets of STIM1 or Orai1-deficient mice showed defective SOCE, but largely intact agonist-induced integrin activation, demonstrating that this process can be initiated by relatively small rises in intracellular Ca2+ transients from the ER.24)25)
Rap1-GTP-interacting adaptor molecule
The Rap1 effector molecule Rap1-GTP-interacting adaptor molecule (RIAM) is a member of the MRL (Mig-10, RIAM, and lamellipodin) family of adaptor proteins.26) MRL proteins function as scaffolds to connect the membrane targeting sequences in Ras GTPases to talin-1. This interaction of MRL proteins with both Rap1-GTP and talin-1 mediates adhesion of β1 and β2 integrins.27) In living cells, RIAM over-expression stimulates talin recruitment to αIIbβ3 with integrin activation,19) but RIAM knockdown blocks talin recruitment to αIIbβ3 with integrin response.28) These findings suggest the important interaction of Rap1-GTP, RIAM and talin-1, resulting in the binding of talin-1 to integrin β cytoplasmic tails.29)
Akt
PI-3K, via phosphatidylinositol-dependent kinase-1, generates phosphoinositide products including Akt, which is a family of intracellular serine/threonine protein kinases (also called protein kinase B).30) Three isoforms of Akt, which possess more than 80% homology, have been identified: Akt1, Akt2, and Akt3. Platelets contain both the Akt1 and Akt2 isoforms. Platelets from Akt1-deficient mice exhibited defects in dense and α-granule secretion, reduced fibrinogen binding, and impaired aggregation in response to low concentrations of thrombin.31) Also, Akt2-deficient mice showed impaired platelet aggregation, fibrinogen binding, and granules secretion especially in response to low concentrations of thrombin and TXA2, which activate the Gq-coupled receptors.30) The above findings showed that both Akt-1 and Akt-2 are important in low dose agonist-induced platelet activation and in platelet-dependent thrombus formation.
Several signaling molecules have been implicated as the downstream targets of PI-3K- and Akt-mediated platelet activation. These include glycogen synthase kinase (GSK)-3β, nitric oxide synthase (NOS) 3, and cAMP-dependent phosphodiesterase (PDE3A). GSK-3β suppresses platelet function and thrombosis in mice. Akt-mediated phosphorylation of GSK-3β inhibits its suppressive function of platelet activation.32) Akt-mediated stimulation of NOS3 induces protein kinase G-dependent degranulation of platelets.33) In addition, Akt has been implicated in activation of PDE3A, resulting in reduced platelet cAMP levels after thrombin stimulation.34)
Talin-1
Talin is one of several proteins that link the cytoplasmic domains of integrin β subunits to actin filaments. Binding of talin to cytoplasmic domains of β-integrins triggers a conformational change in the extracellular domains that increases its affinity for ligands.35)
Talin-1, a 270-kDa cytoplasmic protein, is composed of a globular N-terminal head domain and a flexible rod domain, which can be dissociated by the protease calpain 2. The head region of talin-1 contains a FERM (protein 4.1, ezrin, radixin, moesin) domain comprising 3 subdomains F1, F2, and F3, which has binding sites for the cytoplasmic domains of β-integrins, as well as for filamentous actin (F-actin). The talin rod contains an additional integrin-binding site, at least 2 actin-binding sites and several binding sites for vinculin.36)
The cytoplasmic domains of β-integrins are essential for integrin activation. The β3 tail interacts with a large number of cytosolic proteins to induce the conformational change of the extracellular domain. To identify structural features responsible for these interactions, nuclear magnetic resonance (NMR) studies about β3 transmembrane (TM) and cytoplasmic domains were performed.37)38) The results of these studies showed 3 α helices followed by a short unstructured C-terminus. The first helix, β3 TM helix, embedded in the lipid bilayer, but oriented at a tilted angle because the length of this helix is longer than that of a typical membrane bilayer. The β3 TM helix is followed by a hinge at residues H722-D723, the second helix (residues K725-A735), a hinge containing NPLY motif (residues 744-747), and the third helix at the C-terminal end.
In one suggested model for talin-mediated activation of integrins, the low-affinity αIIbβ3 is maintained through the interaction between α and β subunit at the TM and membrane-proximal domains. The TM helices specifically pack together and are in a long and tilted geometry. The membrane-proximal regions of αIIb and β3 cytoplasmic tails are believed to interact through a salt bridge between a conserved Arg in the αIIb tail and an Asp in the β3 tail, thereby keeping αIIbβ3 in the inactive state. The F3 subdomain of talin FERM domain contains a phosphotyrosine binding (PTB) domain, which interacts with NPLY motif of β3 tail. This interaction is proposed to induce αIIbβ3 activation by disrupting salt bridge formed between αIIb-Arg995 and β3-Asp723, and hence inducing a series of conformational changes that give rise to the affinity modulation of the extracellular domain.19)
The analysis of platelets from conditional talin-1-deficient mice (Tln-/-, CreloxP) supported an essential function of talin-1 in platelet integrin activation.39)40) Talin-1-deficient platelets were unable to activate integrin αIIbβ3 in response to any tested agonists and consequently did not aggregate. Furthermore, they failed to spread on immobilized fibrinogen, suggesting that talin-1 is required for αIIbβ3-dependent outside-in signaling, as well as inside-out signaling. This complete lack of integrin function in Tln-/-platelets resulted in defective hemostasis and thrombus formation in injured vessels in vivo.
Kindlin-3
Kindlins constitute a family of evolutionarily conserved cytoplasmic components of cell-extracellular matrix adhesions that bind to cytoplasmic tails of β-integrins directly and cooperate with talin in integrin activation. There are 3 mammalian kindlins: kindlin-1 (also known as kindlerin and FERMT1), kindlin-2 (also known as MIG-2) and kindlin-3 (also known as URP 2). They exhibit identical domain architecture and high sequence similarities. One of the important features of kindlins is that they contain a FERM domain, which binds to the cytoplasmic tail of β1 and β3 integrins. So far, 2 more kindling binding proteins other than β integrins have been identified: integrin-linked kinase (ILK) and migfilin.41)
It is proposed that the PTB site of the F3 domain in kindlin-3 directly interacts with the NITY motif (residues 756-759) located just after the 3rd helix of integrin β tails.35) Different binding regions of talin-1 and kindlin-3 allow either simultaneous or sequential binding of the 2 proteins to activate the integrin.16)
Platelets from fetal liver cell chimeric kindlin-3-/- mice were unable to activate integrin αIIbβ3 in response to any tested agonist, despite unaltered expression of talin-1, and kindlin-3-/- platelets did not spread on fibrinogen. This complete loss of integrin function resulted in a severe hemostatic defect and resistance to arterial thrombosis in kindlin-3-/-chimeric mice.42)
Platelet Aggregation
The final common step leading to the formation of the platelet plug is platelet aggregation. Platelet aggregation is mediated by the activated GP IIb/IIIa, which binds fibrinogen and von Willebrand factor at high affinity. The dimeric structure of fibrinogen and the multimeric structure of vWF allow these ligands to act as bridges between adjacent platelets and to generate a platelet aggregate.15)
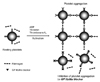
GP IIb/IIIa receptor blockers inhibit fibrinogen binding to the receptors, resulting in the suppression of platelets aggregation (Fig. 5). Abciximab, chimerized monoclonal antibody to integrin αIIbβ3 (c7E3) to reduce immunogenicity, was the first GPIIb/IIIa receptor blocker to be widely used in humans. The peptides recognized by integrin αIIbβ3 were used in drug development. Eptifibatide contains the amino acid sequence, Lys-Gly-Asp (KGD) within a disulphide ring. Tirofiban is a synthetic, non-peptide inhibitor based on RGD sequence.17)18)
Conclusion
Numerous antiplatelet agents have been developed based on the platelet activation mechanism, and some of these agents have been being used for the primary and secondary prevention of cardiovascular diseases. Sometimes dual or triple antiplatelet agents, GP IIb/IIIa antagonists, or more potent drugs, are required to prevent thrombotic complications, especially in patients with acute coronary syndrome or coronary stents. Receptors and signaling proteins involved in platelet activation are targets for a new antiplatelet agent. Through an improved understanding of activation mechanism of platelet and integrin αIIbβ3, more effective antiplatelet therapy can be developed.



 XML Download
XML Download