

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Loeys-Dietz syndrome is a rare genetic syndrome that predisposes patients to aortic aneurysm, and results from mutations in receptors for cytokine transforming growth factor-beta (TGF-ß). It is characterized by a triad of features, including generalized arterial tortuosity, hypertelorism, and a bifid uvula or cleft palate.1)2) It is also associated with a high risk of premature aggressive aortic aneurysm and dissection or rupture at a young age, even when the aortic diameter is small.2)3) We describe the case of an adolescent, who underwent serial near total aortic replacement, from the aortic valve to the descending aorta, with Loeys-Dietz syndrome confirmed by the detection of a mutation in the TGF-ßR 2 gene.
Case
A 15-year-old boy presented to the emergency room (ER) with chest pain that had lasted over 2 hours. The pain started as pulsating back pain, and then transformed into abdominal and chest pain. The pain score was reported to be 7-8 points out of 10.
The patient's family history was unremarkable. He had a complex past medical history and associated anomalies. The patient had suffered from a cleft palate, dolicocephaly, craniofacial dysraphism, and deformity of the first metaphalangeal joint since birth. In the first month of life, he was diagnosed with an enlarged aortic annulus, a dilated pulmonary annulus, a patent ductus arteriosus, and an atrial septal defect, as detected by echocardiography. At 16 months of age, he underwent triple suture ligation of a 6-mm-sized patent ductus arteriosus at our hospital. At 2 years of age, he was admitted to the neurosurgery department because of progressive frontal bossing. Preoperative echocardiography showed a severely dilated aortic sinus (largest diameter: 42 mm) with moderate aortic regurgitation. Aortic valve sparing root replacement using Hemashield (20 mm; Meadox Medical, Inc., NJ, USA) was performed because of an increased aortic sinus with the risk of rupture. Besides cardiovascular surgery, the patient underwent a ventriculo-peritoneal shunt operation because of developing hydrocephalus at 30 months of age, cervical vertebrae laminectomy with rib bone graft, due to occipital C2-3 fusion at 3 years of age, and total calvarial reconstruction to correct dolicocephaly at 8 years of age. When he was 10 years old, composite graft interposition for annuloaortic ectasia (Bentall operation) and aortic valve replacement was performed with the St. Jude regent with a 23-mm graft interposition (St. Jude Medical, Inc., MN, USA), due to aortic annular ectasia and significant aortic regurgitation.
The patient was prescribed warfarin, atenolol, and losartan before his visit to the ER. Sixteen days prior to presenting with chest pain, computed tomographic (CT) angiography during a routine checkup showed diffuse dilatation of his thoraco-abdominal aorta (maximal diameter at the thoraco-abdominal junction, 37 mm).
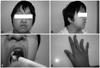
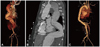
In the ER, the patient's vital signs were as follows: blood pressure, 139/65 mm Hg; pulse rate, 124 beats per minute; respiration rate, 26 breaths per minute; body temperature, 36.7℃. He had an audible aortic valve click at the right upper sternal border, and a palpable pulsating mass around the periumbilical area. A CT angiogram revealed type B aortic dissection, from the descending thoracic aorta to the level of the upper portion of the superior mesenteric artery (maximal diameter at the thoraco-abdominal junction, 42 mm). We maintained atenolol and losartan treatment, and started nitroglycerin infusion with subsequent intravenous nicardipine infusion. This was done instead of an emergency operation because the aortic dissection was type B. On the second day of illness, the dose of losartan and atenolol was increased in order to control the patient's blood pressure. After admission, he occasionally complained of back pain. A follow-up CT angiogram revealed an increased false lumen without ischemia-associated symptoms. On the sixth day of illness, the echocardiogram showed a large aorta with a dissection measuring 44.4×40.3 mm at the diaphragm level, mild mitral regurgitation, and tricuspid regurgitation. We suspected the patient had Loeys-Dietz syndrome, characterized by the triad of arterial tortousity and aneurysm, hypertelorism, and bifid uvula or cleft palate (Fig. 1). Therefore, we studied TGF-ßR 1 and TGF-ßR 2 genes at this stage. A follow-up CT angiogram on the 18th day of illness revealed an increased descending thoracic aorta diameter. A CT angiogram on the 26th day of illness showed that the maximal diameter of the proximal descending thoracic aorta was 56 mm and that of the thoraco-abdominal junction was 56.85 mm, extending from the left subclavian artery ostium to above the renal arteries (Fig. 2A and B). Since the aortic dissection continued to progress despite aggressive antihypertensive management, we completed total aortic graft replacement of the thoraco-abdominal aortic aneurysm with a Vascutek bridge graft (26×10 mm; Vascutek Ltd., MI, USA) and a Vascutek woven graft (Vascutek Ltd., MI, USA) 20 mm on the 29th day of illness, under deep hypothermia and cardiopulmonary bypass (Fig. 2C). Intercostal anastomosis ranging from T 9 to T 11 was done and a separate anastomosis of celiac axis was used with an 8 mm Vascutek graft. Histologic examination of the aortic wall showed thrombi and vascular proliferation with mild infiltration of lymphocytes and eosinophils in the adventitia. The postoperative clinical course was uneventful, and the patient was discharged on postoperative day 16. After a 10-month follow-up period, the patient continues to do well.
Deoxyribonucleic acid sequencing in this patient showed a transition
from thymine to cytosine within the serine-threonine kinase
domain of TGF-ßR 2 (C533R), with identification of a previously
reported missense mutation for Loeys-Dietz syndrome (Fig. 3).
Discussion
We report the case of an adolescent with multiple anomalies requiring several corrective operations, culminating in total artificial valve and graft replacement from the aortic valve to the abdominal aorta except the proximal aortic arch, due to Loeys-Dietz syndrome. Loeys-Dietz syndrome is a recently recognized genetic disorder that presents as aortic aneurysm or dissection, similar to Marfan syndrome. Loeys-Dietz syndrome is subdivided according to the presence (type I) or absence (type II) of craniofacial features such as hypertelorism and bifid uvula.2)3) Type I tends to be associated with more severe cardiovascular anomalies than type II.2)3) Our patient was considered to have type I Loeys-Dietz syndrome, and had a cleft palate and severe aortic aneurismal changes from birth.
Affected patients have a high risk of aortic dissection or rupture from a young age,2)4) and the primary source of early mortality in Loeys-Dietz syndrome is progressive dilatation of the aorta, which is usually maximal at the level of the sinuses of Valsalva. At the time of diagnosis, about two-third of patients with Loeys-Dietz syndrome have an aneurysm of the aortic root, and about one-fifth of patients have an aortic dissection.2)5) The disease progression in our patient was fast, and he underwent aortic surgery twice before he was 10 years old, before finally suffering from aortic dissection and requiring aggressive total aortic graft replacement at 15 years of age.
Current indications for surgery in children with an enlarged aortic root are suggested to be a Z-score of greater than 3.0 or a rapidly expanding aortic root (>0.5 cm over 1 year) in type I Loeys-Dietz patients.3)5) In comparison to Marfan syndrome, aortic aneurysm and dissection occur at smaller aortic diameters in patients with Loeys-Dietz syndrome.
Recent studies recommend a new operative strategy using a specially designed surgical tool or a hybrid aortic arch pair.6) In cases of progressive aortic pathology, open repair of the descending thoraco-abdominal aneurysms may be an appropriate treatment.5)7) On the other hand, the medical management of Loeys-Dietz syndrome, which includes controlling blood pressure, is only symptomatic, and it is impossible to prevent the progression of aortopathy. Patients with Loeys-Dietz syndrome usually undergo multiple cardiovascular operations, and we propose that total aortic replacement would be of benefit, as in the case of our patient.8)
In cases of progressive aortic pathology, timely recognition and aggressive surgical intervention is crucial for proper management.3)5)9) However, preventive surgical repair is still controversial.10) Patients who present with symptoms of Loeys-Dietz syndrome at an early age should undergo meticulous follow-up of their cardiovascular status, through echocardiography and cardiac CT or magnetic resonance imaging (MRI).3)5)

 XML Download
XML Download