

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Autosomal dominant polycystic kidney disease (ADPCKD) has been associated with diverse heart valvulopathies (aortic or atrioventricular valve regurgitation), cerebrovascular aneurysm, and aortic aneurysm. In ADPCKD, aneurysms are most commonly found in the abdominal aorta and generally discovered in patients with advanced renal dysfunction or in patients on maintenance haemodialysis. Aortic root aneurysms and ascending thoracic aortic aneurysm (ATAA) occurring concomitantly with severe aortic regurgitation (AR) is very rare, especially in young patients with normal renal function in ADPCKD. Here we present a case of severe AR caused by marked ATAA associated with ADPCKD in a patient with normal renal function. Surgical intervention was necessary to correct the underlying aortic problem and the associated aortic valve pathology.
Case
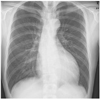
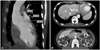
A 27-year-old Asian male with dyspnea upon exertion was referred to our outpatient cardiology clinic. The patient's dyspnea had worsened gradually over the prior 5-6 years and had been exacerbated over the past 3 months. The physical examination result was normal habitus, without Marfan's syndrome phenotypes. His vital signs were as follows: blood pressure, 126/78 mm Hg; pulse rate, 95 beats/minute; respiratory rate, 22 breaths/minute; body temperature: 36.7℃. Both jugular veins were slightly engorged. A high-pitched, blowing, holodiastolic murmur (grade V/VI) was clearly audible at the right sternal border of the third intercostal space. No crepitation or wheezing was observed in his chest, and his abdominal examination was normal. An electrocardiogram showed left ventricular hypertrophy and bi-atrial enlargement in normal sinus rhythm. Chest radiography revealed mild cardiomegaly without pulmonary congestion (Fig. 1). All blood chemistry profiles were within normal limits; they were as follows: fasting glucose 92 mg/dL, total cholesterol 214 mg/dL, creatinine 1.0 g/dL, blood urea nitrogen 21 mg/dL, uric acid 5.7 mg/dL. Echocardiogram showed left ventricular enlargement (end-diastolic dimension, 73 mm), left ventricular hypertrophy (septal wall thickness on diastole, 15 mm), and moderate left ventricular systolic dysfunction with a left ventricular ejection fraction of 36% (Fig. 2A). Dimensions of the sinus of Valsalva and aortic valve opening were 56 mm and 34 mm (Fig. 2B). Morphology of his aortic valve leaflets in a 2 dimensional echocardiogram was normal, but severe aortic valve regurgitation due to aortic root dilation was observed in a Doppler echocardiogram (Fig. 2C). The chest CT confirmed marked aortic root dilation and ATAA (Fig. 3A). Multiple variable-sized cystic lesions in the liver and both kidneys were observed in an abdominal-pelvic CT scan that was compatible with ADPCKD (Fig. 3B). An operation was undertaken involving composite graft replacement of the aortic valve, aortic root and ascending aorta, otherwise known as the Bentall operation. The aortic root was excised and replaced with a 26-mm INTERGARD woven vascular graft (MAQUET GmbH & Co. KG, Rastatt, Germany). An aortic valve replacement was done with a Saint-Jude 25 mm mechanical aortic valve (St. Jude Medical, St. Paul, MN, USA). Both coronary arteries were re-implanted on the side of the graft. During weaning from cardiopulmonary bypass post-operatively, an intra-aortic balloon counter pulsation was inserted for prophylactic reasons against systolic dysfunction. In the pathological examination of the excised aorta, cystic medial necrosis, which had been defined as mucoid material accumulation, was noticed in the media on Hematoxylin and eosin (H & E) staining (Fig. 4A). Necrotized, disorganized smooth muscle cells, and elastic fragmentations were observed in elastic fiber staining (Fig. 4B).1) These findings were compatible with typical pathologic findings of large vessels in ADPCKD.1) The patient's recovery was quick, and it was without incident as far out as 4 years post-operation, with the exception of one small renal cyst rupture. After the operation, the patient has continued to take furosemide, spironolactone, angiotensin-converting enzyme inhibitor, and warfarin. The last measured serum creatinine level was 1.30 mg/dL (upper normal limit, 1.20 mg/dL).
Discussion
Autosomal dominant polycystic kidney disease is characterized by progressive expansion of numerous fluid-filled cysts, resulting in a massive enlargement of the kidneys and eventual extra-renal cyst formation.2) ADPCKD is predominantly a systemic disease seen in adults, resulting from mutations in either the PDK-1 or PDK-2 gene.3) The PDK-1 encoded protein, polycystin-1, is a large receptor-like molecule, whereas the PDK-2 gene product, polycystin-2, has features of an ion-channel protein.3) ADPCKD shows numerous variable-sized cysts in the renal cortex and medulla. It is different from autosomal recessive polycystic kidney disease, which shows small-sized cysts, less than 5 mm diameter, limited to the collecting tubules.2) Feature of imaging modality of this patient was compatible to ADPCKD in the aspect of numerous cysts formation in renal cortex, medulla, and liver. Worldwide, ADPCKD occurs in 1 : 400 to 1 : 1000 individuals and can lead to ESRD; the annual U.S. incidence is 6.0-8.7 per million.2)3) Diverse extra-renal manifestations of ADPCKD highlight the systemic nature of the disease and likely reflect a generalized abnormality in both collagen content and the composition of the extracellular matrix.4) With this pathologic background, ADPCKD can lead to abnormalities in vessel walls and cardiac valve disorders, and 25% of ADPCKD patients suffer from cardiac valvulopathies such as prolapse or regurgitation in the atrioventricular valves or aortic valve regurgitation.3)5) In a combined retrospective and prospective study, 11 patients with ADPCKD were found with one or more cardiac or aortic lesions.5) Seven patients had primary dilation of the aortic root and annulus with AR. The severity of the AR necessitated aortic valve replacement in 2. Mitral regurgitation was present in 3 patients, of whom 2 had documented redundant mitral leaflets and ruptured chordate tendinae, and the third had mitral valve prolapse. Histological analysis of aortic and mitral valve tissue from these acquired lesions showed myxomatous degeneration with loss and disruption of collagen.5) The most common cause of death in 129 studied ADPCKD patients receiving haemodialysis was cardiac disease, and most autopsy studies of ADPCKD patients uncovered cardiac hypertrophy and coronary artery disease.6) To date, there are a limited number of case reports documenting abdominal aortic aneurysm (AAA) and aortic diameter increment compared to glomerulonephritis in ADPCKD.7)8) Of the reported cases involving AAA and its rupturing or dissection in ADPCKD, almost all patients were elderly with long-standing complications such as hypertension or advanced renal insufficiency or associated haemodialysis.9) Autopsy analysis revealed dissections of the thoracic aorta to be 7 times more common in ADPCKD patients than in normal populations, but cases of thoracic dissection or TAA that require surgery are rarely found in living ADPCKD patients.10)11) Therefore, a case of severe AR needing surgical correction to address an ATAA caused by ADPCKD in a young patient - especially one having normal renal function - is rare and is not found in the medical literature. Our patient had neither Marfan's syndrome habitus nor risk factors for atherosclerosis, e.g., prolonged hypertension, diabetes mellitus, smoking, or dyslipidemia. In pre-operative chest and abdominal-pelvic CT imaging, we found numerous variable-sized cysts compatible with ADPCKD in the renal medulla, renal cortex and liver. Based on the progressive severe dyspnea and imaging confirmation of aortic pathology, surgery was performed. This involved the aortic root being surgically removed and replaced with a synthetic vascular graft, and an aortic valve was also replaced with a mechanical valve. In the surgical specimen of the aorta, we observed cystic medial necrosis on H & E staining (Fig. 4A), and this phenomenon was confirmed with necrosis, disorganization of smooth muscle cells, and fragmentation of elastin in elastic fiber staining (Fig. 4B). With these remarkable characteristics in the aorta, we surmised a pathological relationship between ADPCKD and ascending aortic dilation and AR.1) After the operation, magnetic resonance imaging and angiography for the brain were performed to rule out the existence of cerebral arterial aneurysm. Fortunately, this patient had no evidence of a cerebral aneurysm or any other vascular abnormality. We planned regular surveillance for cerebral aneurysm and renal function deterioration. Because the ADPCKD patients have more risk on an anticoagulation therapy, for example, life-threatening renal cyst bleeding, or cerebral aneurismal bleeding, the careful monitoring of anticoagulation intensity, and continuous teaching for this risk on patients should be considered.12) Even though the mechanistic association between ADPCKD and aortic aneurysm is not clear, ADPCKD should be taken into account as a possible cause of severe AR related with ascending ATAA in cases that have no identifiable causes of aortic atherosclerosis or cystic medial necrosis, especially at younger ages.3)6)


 XML Download
XML Download