

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) syndrome is a multisystem disorder, which is clinically characterized by encephalopathy, frequently manifesting as dementia, seizures and stroke-like episodes at a young age with evidence of mitochondrial dysfunction in the form of lactic acidosis and ragged-red fibers. Multiple organs can be affected, but tissues with high energy demand are the most vulnerable. Due to the high energy requirement of cardiac muscles, cardiac disease features prominently among the systemic manifestations of MELAS syndrome. Although cardiomyopathy is the most common cardiac manifestation, Wolff-Parkinson-White (WPW) syndrome has also been reported in association with MELAS syndrome. We report a case of a WPW syndrome in a patient with MELAS syndrome.
Case
A 21-year-old woman was admitted to the hospital for a seizure-like episode lasting for approximately five minutes and subsiding spontaneously. The patient had frequent and insidious onset of seizure-like episodes, dysarthria, gait disturbance and a right-sided visual field defect that had started four years ago without any history of essential hypertension, diabetes mellitus and dyslipidemia.
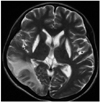
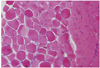
The patient initially presented with blood pressure of 111/59 mm Hg, pulse rate of 109 beats/min, respiratory rate of 20/min, and body temperature of 36.0℃. Laboratory results were raised cerebrospinal fluid (CSF) lactate of 5.2 mmol/L. Electroencephalography (EEG) showed diffuse cerebral dysfunction. Electromyography revealed sensorimotor polyneuropathy and chronic myopathy. Brain MRI showed infarction in the right temporal lobe, ischemia in the left posterior frontoparietal cortex and basal ganglia, and cystic lesion in the pineal gland with brainstem and cerebellar atrophy (Fig. 1). Biopsy of left vastus lateralis showed neurogenic atrophy and slightly increased lipid vacuoles without paracrystalline inclusion in the mitochondria (Fig. 2). Based on these clinical findings, the patient was diagnosed with MELAS syndrome.
Ten years later, she went to a dentist for treatment of dental caries. Five minutes after local anesthesia with lidocaine, her consciousness changed suddenly and she was urgently transferred to the emergency room of our institution.
On arrival, she was stuporous with blood pressure of 209/147 mm Hg, pulse rate of 140 beats/min, respiratory rate of 25/min, and body temperature of 36.5℃. At that time, her electrocardiography (ECG) showed supraventricular tachycardia. After intravenous injections of 300 mg amiodarone, she recovered consciousness, and follow-up ECG showed sinus rhythm with pre-excitation (Fig. 3).
Transthoracic echocardiography revealed decreased early diastolic mitral annulus velocity (E' velocity) and abnormal myocardial texture which were possibly associated with the initial phase of restrictive cardiomyopathy. Considering her bedridden status due to underlying MELAS syndrome, medical treatment with propafenone was started. The patient was discharged and has been followed up without tachycardia attack.
Discussion
To the best of our knowledge, this is the first case to report on WPW syndrome in a patient with MELAS syndrome in the Korean literature. MELAS syndrome is a maternally inherited disorder caused by mutations of mitochondrial DNA that presents with a wide range of clinical expression. MELAS syndrome is clinically characterized by 1) encephalopathy, frequently manifesting as dementia, seizures, or both; 2) stroke-like episodes at a young age (typically by age 40 years); and 3) evidence of mitochondrial dysfunction in the form of lactic acidosis, ragged-red fibers or both.1)
Age at onset was initially described to range from under 2 years to over 60 years.2) Multiple organs can be affected, but tissues with high energy demand are the most vulnerable. Therefore, nervous system and muscle impairment often dominate the clinical picture.3) Cardiomyopathy is an early recognized cardiac manifestation and is now a widely accepted complication of MELAS syndrome. Kim et al.4) reported the first case of myocardial involvement in a patient with MELAS syndrome in the Korean literature, and Son et al.5) also reported a case of myocardial involvement in a patient with mitochondrial myopathy.
Conduction defects, including WPW syndrome, have also been reported in association with MELAS syndrome.6) Recently, Sproule et al.7) reported that four (13%) of 30 patients with MELAS syndrome had a clinical history of, or ECG findings consistent with, WPW syndrome.
Laboratory evaluation is a logical initial step in the diagnostic evaluation of suspected MELAS syndrome and an elevated level of lactate in the CSF is helpful. MRI is a powerful tool in the diagnostic evaluation of suspected cases of MELAS syndrome. The Brain MRI of patients with MELAS syndrome typically demonstrates asymmetric lesions of the occipital and parietal lobes that mimic ischemia.1)3) Muscle biopsy can be vital in the evaluation of MELAS syndrome. The presence of ragged red fibers on muscle biopsy is a classical finding and scattered vacuolated muscle fibers with a clear surrounding rim as well as the presence of many basophilic inclusions on microscopic examination.6)
The percentage of heteroplasmy for a mitochondrial RNA mutation may vary widely depending on the specific tissue sampled.8) Blood leukocyte samples have traditionally been the most common specimens, and urine sediment, cheek mucosa and skin fibroblasts are often used and easily accessible alternate specimens.9) Although the m.3243A>G common MELAS mutation is responsible for more than 80% of reported MELAS cases,1) most of these reports have come from Western countries. Abu-Amero et al.10) reported the case of a patient with typical clinical features of MELAS syndrome, but mutations in recognized genes were not identified.
Despite considerable advances in genetics and molecular biology, therapeutic options for MELAS syndrome remain limited. Primary current treatment involves symptomatic management, in addition to supplementation with a so-called "mitochondrial cocktail". Although recent studies of a potential therapeutic role for L-arginine have been promising, these observations needs to be confirmed by controlled trials.6)
Because of significant morbidity and mortality, patients with MELAS syndrome should be screened at the time of diagnosis and followed regularly for the possibility of concurrent cardiac complications such as cardiomyopathy and WPW syndrome.

 XML Download
XML Download