

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The sinoatrial node (SAN) automaticity is responsible for initiating the heart rhythm. So, the SAN function is essential for normal cardiac physiology. Sick sinus syndrome is an abnormality involving the generation of the action potential by the SAN and this is characterized by an atrial rate that is inappropriate for the physiological requirements. Sick sinus syndrome occurs in 1 of every 600 cardiac patients older than 65 years and it accounts for approximately half of the implantations of pacemakers in the United States.1)
The spontaneous firing of a subsidiary pacemaker (SP) is important as the secondary pacemaker in patients with sick sinus syndrome. Randall et al.2) reported that a supraventricular (atrial) rhythm resumed after surgical excision of the SAN in both resting and exercising dogs. Waxing and waning of the heart rate, which is generally referred to as sinus arrhythmia, continues in the awake animal after complete excision of the SAN. This arrhythmia is responsive to both adrenergic and cholinergic interventions, and this indicates the presence of both sympathetic and parasympathetic regulation of the subsidiary atrial pacemaking tissues.2) Ligation of the SAN artery at the mid portion of the sulcus terminalis suppressed the SAN activity and elicited SP activity in a well-defined region of the inferior atrium.3)
Spontaneous diastolic depolarization of the SAN cells periodically initiates action potentials to set the rhythm of the heart. The mechanism of spontaneous diastolic depolarization has traditionally been attributed to a "voltage clock" mechanism that is mediated by voltage-sensitive membrane currents such as the hyperpolarization-activated pacemaker current (If) regulated by cyclic adenosine mono-phosphate.4)5) The "Ca2+ clock" has recently been proposed as a complimentary mechanism of SAN automaticity.6-14) The Ca2+ clock is mediated by rhythmic spontaneous sarcoplasmic reticulum (SR) Ca2+ release, which in turn activates the Na+/Ca2+ exchanger current (INCX) and causes diastolic depolarization.6)8-11)15) However, the mechanism of spontaneous firing of the SP has not been revealed. It is also unclear how the heart rate accelerates after sympathetic stimulation of the SP. The purpose of the present study was to make various pacemaker models and reveal the mechanism of spontaneous firing and heart rate acceleration by sympathetic stimulation of the SP.
Materials and Methods
Various Langendorff-perfused canine right atrium preparations
This study's protocol was approved by the Institutional Animal Care and Use Committee and it conforms to the guidelines of the American Heart Association. We studied the isolated canine right atriums (RA) from 15 mongrel dogs (22 to 28 kg). The dogs were intubated and anesthetized with isoflurane. The chest was opened via a median sternotomy and the heart was rapidly excised. The dogs were euthanized by exsanguination during general anesthesia. The right coronary artery was perfused with 37℃ Tyrode's solution equilibrated with 95% O2 and 5% CO2 to maintain a pH of 7.4. The composition of Tyrode's solution was (in mmol/L): 125 NaCl, 4.5 KCl, 0.25 MgCl2, 24 NaHCO3, 1.8 NaH2PO4, 1.8 CaCl2 and 5.5 glucose. The perfusion pressure was between 50 mm Hg and 60 mm Hg. All the ventricular coronary branches were tied off. Both ventricles and the left atrium were excised and removed from the preparation. During the optical recordings, contractility was inhibited by 10-17 µmol/L of blebbistatin.12)
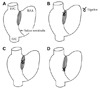
We produced various RA perfusion models by isoproterenol infusion and ligation of the SAN artery. The superior RA model was produced by isoproterenol infusion of 1 µmol/L. The middle and inferior SAN models were produced by the ligation of the sulcus terminalis at the upper sixth portion and the upper third portion. Finally, the SP model was generated by ligating the mid portion of the sulcus terminalis (Fig. 1). The time interval between the each ligation was 20 minutes.
Dual Vm and Cai recordings
Dual Vm and Cai recording was performed in 9 dogs. We used 0.5 mg of Rhod-2 AM (Molecular Probes) dissolved in 1 mL of dimethylsulfoxide containing Pluronic F-127 (20%wt/vol) to stain the Cai. This solution was diluted in 300 mL of Tyrode's solution to achieve a final Rhod-2 concentration of 1.48 µmol/L and this was infused into the heart over a 10 minutes period. The heart was perfused with dye-free Tyrode's solution for 15-30 minutes for de-esterification of Rhod-2 AM. The hearts were then stained again by direct injection of voltage sensitive dye (RH237, Molecular Probes) into the perfusion system.12) The double-stained hearts were excited with two LED lamps at 532 nm. The fluorescence was collected using a 2 image-intensified charge-coupled device (CCD) camera (Dalsa Inc., Waterloo, Canada) at 3.75 ms/frame and 125×125 pixels with spatial resolution of 0.24×0.24 mm2/pixel.16) The CCD camera for the Vm was fitted with a 715-nm long-pass filter, and the CCD camera for the Cai was fitted with a 580±20-nm band-pass filter. After dual Vm and Cai optical mapping of the baseline spontaneous beats, isoproterenol was infused starting from 0.01 µmol/L and this was gradually increased to 1.0 µmol/L. The isoproterenol dose responses were evaluated in 6 RAs with intact SANs and 3 SP models. Because late diastolic Cai elevation (LDCAE) was not observed in all 3 SP models, Vm recordings were performed in 6 additional SP models using di-4-ANEPPS (Invitrogen, California, USA). The pharmacologic interventions were as follows: ZD 7288 (3 µmol/L, n=2), ryanodine (3 µmol/L, n=2), isoproterenol (1 µmol/L, n=6) and then this was followed by ZD 7288 (3 µmol/L, n=3), ryanodine (3 µmol/L, n=2) and ryanodine (10 µmol/L) plus thapsigargin (200 nmol/L) (n=3). Finally, to block both the Ca2+ and voltage clocks, a combined infusion of ryanodine (10 µmol/L), thapsigargin (200 nmol/L) and ZD 7288 (3 µmol/L) was performed in 3 SP models.
Data analysis
The Cai and Vm traces were normalized to their respective peak-to-peak amplitude for comparison of the timing and the morphology. We generated activation isochronal maps using the onset of the phase 0 of the action potential in the leading pacemaker site as time 0. The time of the isochronal map is the time when the dVm/dt reached the peak amplitude of phase 0. Student's t-tests were used to compare means between the two groups. A p<0.05 was considered significant.
Results
The effects of sequential ligation of the sinoatrial node artery on the heart rate and the pacemaking site
The SAN artery was ligated sequentially at the superior to middle portion of the sulcus terminalis. Fig. 2 shows the differential responses of the different SAN models to isoproterenol infusion. At baseline, the mean heart rate was 90±23 bpm, and the middle part of the SAN served as the pacemaking site (site 1 of Fig. 2A). After infusing µmol/L of isoproterenol, the heart rate was increased to 189±11 bpm and the leading pacemaking site was shifted cranially and this produced the superior SAN model (site 2 of Fig. 2A). After ligation of the SAN at the superior sixth portion of the sulcus terminalis, the heart rate was decreased to 151±28 bpm and this produced the middle SAN model. The heart rate was 128±26 and 106±11 bpm in the inferior and SP models, respectively (Fig. 2B).
Impaired heart rate response to β-adrenergic stimulation in the subsidiary pacemaker model
Fig. 3 shows the isoproterenol-dose response curve of the heart rate obtained from 6 RAs with an intact SAN and 3 SP models. The baseline heart rate of the SP models was 55±10 bpm, and this was significantly lower than that of the RAs with an intact SAN (90±23 bpm, p=0.004). In the RAs with an intact SAN, an isoproterenol dosage of 1 µmol/L increased the heart rate to 166±18 bpm (102%). In contrast, the heart rate of the SP models increased only to 97±35 bpm (73%, p=0.005) during isoproterenol infusion. Compared with the RAs with an intact SAN, the isoproterenol heart rate response curve of the SP model was shifted rightward. This finding suggests impaired heart rate acceleration at the each dosage of isoproterenol.
The Cai dynamics of the subsidiary pacemaker site after β-adrenergic stimulation
The superior SAN model was produced by infusion of 1 µmol/L isoproterenol in the RAs with an intact SAN. Fig. 4Ba shows the activation map during spontaneous sinus rhythm. The earliest activation site was in the anteroinferior region of the SAN (asterisk). Isoproterenol infusion resulted in an upward (cranial) shift of the earliest activation site to the superior SAN (site s, Fig. 4A), and this was coincident with the appearance of robust LDCAEs (arrows in Fig. 4B) in this region in all 6 preparations.
However, LDCAEs were not observed in the SP model. Fig. 5Ba shows the activation map during spontaneous rhythm in a SP model. The earliest activation site was located below the ligation site at the inferior portion of the sulcus terminalis. Fig. 5Bb shows the activation map during 1 µmol/L infusion of isoproterenol infusion. The heart rate increased from 60 bpm to 100 bpm, and the activation site was slighted moved to an anterior adjacent site. However, the Cai tracing showed no LDCAE anywhere in the mapped region (Fig. 5Bb). In addition, the LDCAE was not observed with different dosages of isoproterenol from 0.01 to 10 µmol/L. The ligation of the SAN artery transiently induced an irregular heart beat (Fig. 6B). However, the heart rhythm was stabilized usually 10 minutes after ligation of the SNA artery (Fig. 6C). The activation maps are presented in Fig. 6E.
The effects of sarcoplasmic reticulum inhibition and if blockade on the heart rate and its acceleration by isoproterenol
We could not find the LDCAE in the SP model at baseline and even during isoproterenol infusion. In the state without isoproterenol pretreatment, ryanodine infusion 3 µmol/L decreased the heart rate of the SP from 55±10 bpm to 52±5 bpm (-7% from baseline).
Fig. 6D and F show the Vm tracing and activation map after ryanodine infusion in the state with isoproterenol pretreatment. The leading pacemaker site was shifted from the anterior RA (site b) to the IVC (site c) after ryanodine infusion. Ryanodine also decreased the isoproterenol-induced heart rate increase of the SP from 106±11 bpm (92% from baseline) to 83±3 bpm (50% from baseline) (Fig. 7) in 2 SP models. To fully suppress the SR Ca2+ cycling, we also studied the effects of ryanodine (10 µmol/L) in combination with thapsigargin (200 nmol/L). After 30 minutes treatment, the mean heart rate further decreased to 69±7 bpm (24% from baseline, n=3).
In contrast to the SR inhibitors, the If blocker ZD 7288 (3 µmol/L) decreased the basal heart rate to 44±5 bpm (-21%, n=2), and it also partially prevented isoproterenol from increasing the heart rate to 74±5 bpm (33% from baseline, n=3) (Fig. 7).
If the voltage and Cai clocks act interdependently and synergistically to support each other in determining the SP rate, then the effects of blocking one clock or the other are likely to be complex and not necessarily very informative. This was the case, as shown in Fig. 7, which summarizes the relative changes in heart rate caused by various pharmacologic agents under basal conditions and during isoproterenol infusion. In addition, the isoproterenol-induced heart rate increase was completely abolished by combined funny current blocker and SR Ca2+ emptying. The combined infusion of ryanodine (10 µmol/L), thapsigargin (200 nmol/L) and ZD 7288 (3 µmol/L) further decreased the heart rate to 44±10 bpm (-20% from baseline, n=3) (Fig. 7).
Discussion
Major findings
The present study supports the importance of the If current and spontaneous SR Ca2+ release to the pacemaking of the SP. The key findings include the demonstration of various pacemaker models with different pacemaking sites and heart rates. The block of spontaneous SR Ca2+ release decreased the firing rate of the SP, and this showed the concomitant suppression of heart rate acceleration by β-adrenergic stimulation. The baseline heart rate and heart rate acceleration by β-adrenergic stimulation was partially blocked by ZD 7288, which is a specific If current inhibitor. In addition, we have demonstrated that the isoproterenol-induced heart rate acceleration was completely abolished by blocking both the If current and the spontaneous SR Ca2+ release. Together, these findings support the conclusion that spontaneous SR Ca2+ release acts synergistically with the activation of membrane ionic currents such as If, to accelerate the heart rate in a SP model during β-adrenergic stimulation.
Subsidiary pacemaker models
Rozanski et al.3) suggested that ligation of the SAN artery at the mid portion of the sulcus terminalis suppressed SAN activity and it usually (73.5%) elicited ectopic atrial rhythm activity in a well-defined region of the inferior atrium. In this study, we sequentially ligated the SAN artery from the upper part to the mid portion of the sulcus terminalis, and we confirmed the decrease of heart rate and shifting of the leading pacemaker sites. The pacemaker site was well correlated with the heart rate. In this study, after the ligation of the SAN artery at the upper sixth of the sulcus terminalis, the heart rate was not increased to the level of the superior SAN model and the leading pacemaker site was the middle part of the SAN. Fedorov et al.17) suggested that the canine SAN is insulated from the surrounding atrial myocardium except for 2 narrow superior and inferior sinoatrial exit pathways. In this case, the superior exit was blocked first because we ligated the SAN artery from the superior portion of the sulcus terminalis. However, sequential ligation of the SAN artery always decreased the heart rate. Therefore, our study suggests that the pacemaker hierarchy is the shifting of the leading pacemaker site in the SAN itself rather than the change of the exit site.17)18) Our result supports the pacemaker hierarchy suggested by Boineau et al.19)20)
Mechanisms of rhythm generation of the subsidiary pacemaker
Multiple time- and voltage-dependent ionic currents have been identified in cardiac pacemaker cells and these currents contribute to diastolic depolarization, including If, ICa-L, ICa-T, IST and various types of delayed rectifier K currents.4)5)21) The "Ca2+ clock" has recently been proposed as a complimentary mechanism of SAN automaticity.6-11)The mechanism of heart rhythm generation by the SP has not been completely revealed. Using intracellular recordings, Rubenstein and Lipsius.22) suggested that multiple mechanisms participate in the SP automaticity. They included a cesium-sensitive component, a component mediated via the calcium released from the SR and possibly a direct contribution by the slow inward calcium current. However, the cells in different portions of the RA exhibit a range of electrophysiological and Ca2+ handling characteristics.23-26) Moreover, because the leading pacemaker sites are shifting according to sympathetic stimulation, single cell recordings cannot reflect the exact cells from the leading pacemaking site.
Optical mapping of the intact RA to study the SAN function has the advantage that differential responses to β-adrenergic stimulation and other interventions can be characterized regionally within the leading pacemaker site and then compared to the RA. In this study, LDCAE was not observed at base-line and even during isoproterenol infusion in the SP model, while it was observed in the SAN during β-adrenergic stimulation. One possible explanation of this finding is that the Ca2+ clock lagged behind the voltage clock in regulating diastolic depolarization under basal conditions. Alternatively, because each pixel in an optical map contains information from multiple cells, the effects of spatial averaging might have prevented us from documenting smaller spontaneous SR Ca2+ releases or Ca2+ releases from individual SAN cells. The importance of SR Ca2+ release on heart rate acceleration during β-adrenergic stimulation is also supported by the finding that ryanodine and ryanodine plus thapsigargin decreased the heart rate during isoproterenol infusion.
Interdependency of two clocks in the rhythm generation of the subsidiary pacemaker
The interdependence and synergy between the two clocks is evident from the pharmacologic responses (Fig. 7). If the two clocks were independent with the faster clock driving the heart rate under a given condition, then blocking the slower clock should have no effect on heart rate, whereas blocking the faster clock should decrease the heart rate to that of the slower clock (which could be either markedly or only slightly slower than the faster clock). However, since both SR inhibitors and If blockade slowed the heart rate under basal conditions, as well as this blunted the isoproterenol-induced increase in the heart rate, the hypothesis of independent clocks can be excluded. However, as the two clocks are interdependent, the relative potency of SR inhibition versus If blockade on slowing the heart rate (Fig. 7) becomes very difficult to interpret since interdependency implies that inhibiting the SR Ca2+ release will affect the voltage clock as well as the Ca2+ clock, and vice versa. The interpretation is further complicated by the presence of ectopic foci outside the mapped region during SAN suppression.
Study limitations
In this study, the level of Cai elevation at the subsidiary pacemaking site was too small to be detected by optical mapping. However, there is no doubt that the SR Ca2+ release contributes in the heart rate acceleration during β-adrenergic stimulation because the heart rate was reduced by ryanodine and a combined infusion of ryanodine and thapsigargin. Pharmacologic agents such as ryanodine, thapsigargin and ZD 7288 are not completely selective, and nonspecific effects cannot be excluded.
Conclusion
The block of spontaneous SR Ca2+ release or the funny current decreased the firing rate of the SP and it showed the concomitant suppression of heart rate acceleration by β-adrenergic stimulation. Our results support the conclusion that spontaneous SR Ca2+ release acts synergistically with the activation of membrane ionic currents such as If, to accelerate the heart rate in the SP model during β-adrenergic stimulation.






 XML Download
XML Download