

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The mechanism underlying in-stent restenosis after stent implantation has been shown to principally involve neointimal hyperplasia.1-5) It has been reported that there is a strong correlation between the degree of vascular inflammation and neointimal formation.6)7) The pathologic process of instent restenosis is characterized by an inflammatory healing process after stretch and damage of the vessel wall.7-9)
The renin-angiotensin-aldosterone system has been implicated in the pathogenesis of neointimal hyperplasia, and a role for angiotensin II in the migration and proliferation of vascular smooth muscle cells in restenotic lesions has been proposed.10) Moreover, administration of angiotensin converting enzyme inhibitors has been reported to reverse impaired endothelium-dependent responses to acetylcholine in patients with heart failure11) and hypertension,12) and to improve coronary endothelial function after treatment with an angiotensin converting enzyme inhibitor in patients with coronary artery disease.13)
Our hypothesis in the present study was that the angiotensin-converting enzyme inhibitor ramiprilat-coated stent is effective in reducing neointimal hyperplasia and improving en-dothelial function. We examined the anti-proliferative and anti-inflammatory effects of ramiprilat-coated stents in a porcine coronary overstretch restenosis model.
Subjects and Methods
Animal study protocol
This animal study was approved by the Ethical Committee of the Chonnam National University Hospital in Gwangju, Korea. The study animals were female swine weighing 22-35 kg. To decrease acute thrombosis after stenting, pre-medication with aspirin (100 mg) and clopidogrel (75 mg) per day was given for 5 days before the procedure. On the procedure day, pigs were anesthetized with ketamine (20 mg/kg intramuscularly) and xylazine (2 mg/kg intramuscularly). The pigs received 3 liters/min of supplemental oxygen continuously through an oxygen mask. After 2% lidocaine was administered subcutaneously at the cut-down site, the left carotid artery was surgically exposed, and a 7F sheath was inserted. Continuous hemodynamic and surface electrocardiographic monitoring was maintained throughout the procedure. After heparin (5,000 uints) was administered intravenously as a bolus prior to the procedure, the target coronary artery was engaged using standard 7F guiding catheters and control angiograms of both coronary arteries were performed using non-ionic contrast agent in two orthogonal views.
Coating method of ramiprilat into stents
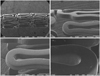
A plasma polymerization reaction was performed to attach an amine radical to the surface of MAC stents (AMG, Munich, Germany). A stent was fixed in a tubular reactor, which was made from a pyrex glass tube, then the pressure was dropped to <5 mtorr by vacuum pump. For attachment of the amine radical to the stent surface, a diaminocyclohexane monomer was drifted to the tubular reactor at a constant dose (0.96 SCCM), and plasma was generated using a radiofrequency power generator. The power for polymerization of plasma was 100 W for 5 minutes, then 60 W for 15 minutes. A glass tube (7 cm in length and 1 cm in width) was boiled in water at 100℃ for 5 minutes, then taken out and allowed to dry in an incubator. Two mL of ramiprilat solution was delivered to the glass tube, then the stent was immersed in the ramiprilat solution. The carboxy radical of ramiprilat was introduced to the amine radical attached to the stent to form a covalent bond, and thus improved the attachment power between the stent and ramiprilat. This reaction was performed for 1 hour, then the stent was taken out and allowed to dry for >24 hours. For release kinetics of ramiprilat from the stent, the stent was placed in a glass vial and immersed in 100 mL of phosphate-buffered saline. The amount of coating of ramiprilat onto the surface of the stent was 90 µg/stent, and the median thickness of coating was 1 µm. The amount of ramiprilat released to the buffer solution was measured using an absorbance test for ultraviolet at 278 nm (Fig. 1). The ramiprilat coating on the surface of the stent was confirmed by scanning electron microscopy (Fig. 2).
Stent-induced stenosis
Pigs were randomized into two groups in which the coronary arteries {16 pigs (16 coronaries in each group)} had a 3.0×17-mm ramiprilat-coated MAC stent or a 3.0×17-mm control MAC stent implanted for proximal epicardial coronary arteries. The stent was deployed by inflating the balloon to nominal pressure at the injury site and the resulting stent-to-artery ratio was 1.3 : 1. Repeat angiograms were obtained immediately after stent implantation. Then, all equipment was removed and the carotid artery was ligated. All animals received 100 mg of aspirin and 75 mg of clopidogrel orally each day until death. Twenty-eight days after stenting, the animals underwent repeat angiography in the same orthogonal views before death and a 20 mL intracoronary injection of potassium chloride was administered. The hearts were removed, and the coronary arteries were pressure-perfusion fixed at 70 mmHg in 10% neutral buffered formalin overnight. The arteries were step-sectioned, and processed routinely for light microscopy.
Histopathologic analysis
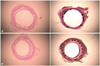
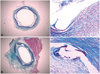
Histopathologic evaluation of each artery was performed by an experienced cardiovascular pathologist. The specimens were embedded in methylmethacrylate and sections were cut with a low-speed diamond wafer mounted to a Buehler Isomet saw (Buehler Ltd., Lake Bulff, IL, USA) leaving the stent wires intact in the cross sections to minimize potential artifacts from removal of the stent wires. Fifty to 100 µm sections were obtained approximately 1 mm apart and stained with hematoxylin-eosin for histologic analysis (Fig. 3). Measurements of the histopathologic sections were performed using a calibrated microscope, digital video imaging system, and microcomputer program (Visus 2000 Visual Image Analysis System, IMT Tech, CA, USA) Borders were manually traced for lumen area, area circumscribed by the internal elastic lamina, and the innermost border of the external elastic lamina (external elastic lamina area). Morphometric analysis of the neointimal area for a given vessel was calculated as the measured internal elastic lamina area minus the lumen area. The measurements were made on five cross sections from the proximal and distal ends and the three midpoints of each stented segments. Histopathologic stenosis was calculated as 100× {1-(lesion lumen area/lesion internal elastic lamina area)}. Fi-brin was identified on hematoxylin and eosin-and Carstair's histochemical-stained sections (Fig. 4).
Evaluation of arterial injury
Arterial injuries at each strut site were determined by the anatomic structures penetrated by each strut. A numeric value was assigned, as previously described by Schwartz et al.14) 0=no injury; 1=break in the internal elastic membrane; 2=perforation of the media; and 3=perforation of the external elastic membrane to the adventitia. The average injury score for each segment was calculated by dividing the sum of the injury scores by the total number of struts at the examined section.
Evaluation of inflammation and fibrin scores
With respect to the inflammation score for each individual strut, the grading was as follows: 0=no inflammatory cells surrounding the strut; 1=light, non-circumferential lymphohistiocytic infiltrate surrounding the strut; 2=localized, moderate-to-dense cellular aggregate surrounding the strut non-circumferentially; and 3=circumferential dense lymphohis-tiocytic cell infiltration of the strut. The inflammation score for each cross section was calculated by dividing the sum of the individual inflammation scores by the total number of struts at the examined section. Inflammatory cell counts were normalized to the injury score in the neointima. The number of uncovered struts (defined as no coverage by any tissue, except the thrombus or fibrin-platelet complex) was recorded. Arterial healing was assessed by fibrin deposition and ordinal data for fibrin were collected on each stent section using a scale of 0-3, as previously reported.15)
Statistical analysis
The Statistical Package for the Social Sciences (SPSS) for Windows (version 15.0; SPSS Inc., Chicago, IL, USA) was used for all analyses. Continuous variables were presented as the mean value±SD; comparisons were conducted by Student's t-test or the Wilcoxon rank-sum test if the normality assumption was violated. Pearson's correlation coefficient was used to determined the correlations between the inflammatory cell counts and the neointimal area, and between the inflammatory cell counts and the percent area stenosis. A value of p<0.05 was considered statistically significant.
Results
Procedural characteristics
One 3.0×17 mm ramiprilat-coated MAC stent or one 3.0×17 mm control MAC stent were placed for 1 epicardial coronary artery per pig. A total of 32 stents were placed in the proximal left anterior descending, proximal left circumflex, and proximal right coronary arteries for 32 pigs (Table 1). The mortality for this study was zero. Quantitative coronary angiography before and after stent implantation showed the stent-to-artery diameter ratio to be 1.323±0.089. There was no significant difference in the stent-to-artery ratio between the ramiprilat-coated control bare-metal stent groups.
Injury and inflammation scores
There were no significant differences in injury and inflammation scores between the ramiprilat-coated and control bare-metal stent groups (Table 2). Within the neointima, most inflammatory cells were lymphohistiocytes. Significant positive correlations existed between the inflammatory counts and the neointima area and between the inflammatory counts and the percent area stenosis (Fig. 5).
Inflammatory reaction, fibrin score, and neointimal reaction
There was no significant difference in the inflammatory cell counts normalized to the injury score between the ramiprilat-coated and control bare-metal stent groups (Fig. 6). There was no significant difference in fibrin scores between the ramiprilat-coated and control bare-metal stent groups (Fig. 7). There was a trend toward a smaller neointima area and a smaller percent area stenosis in the ramiprilat-coated stent group compared with the control bare-metal stent group (Table 2). Uncovered struts did not exist in the ramiprilat-coated and control bare-metal stent groups.
Discussion
This study demonstrated that ramiprilat-coated stents did not have significant inhibitory effects on neointimal hyperplasia; however, ramiprilat-coated stents had good effects on the inflammatory reaction and arterial healing similar to control stents in a porcine coronary restenosis model.
Drug-eluting stents were developed in an effort to reduce the rate of restenosis. Anti-restenotic drugs (sirolimus or paclitaxel) are released over a period as short as days to as long as 1 year after implantation to reduce the local proliferative healing response. However, previous studies have suggested that drug-eluting stents affect poor endothelialization16)17) and delayed arterial healing, which are associated with endothelial dysfunction and inflammation leading to stent thrombosis.16)18)19) The endothelium is a single layer of endothelial cells lining the vascular wall and plays an integral part in maintaining vascular homeostatsis. However, stenting causes significant injury to the vascular wall and endothelium resulting in a proliferative response of the vessel wall and the development of neointimal hyperplasia, and this phenomenon correlates in humans with the plasma angiotensin-converting enzyme concentration.20)21) Furthermore, tissue angiotensin-converting enzyme is up-regulated locally during the vascular healing process.22) Angiotensin-converting enzyme inhibition is thought to potentially contribute to the prevention of tissue angiotensin-converting enzyme up-regulation after stent implantation.
Stimulation of angiotensin II type 1 receptors after arterial injury during balloon angioplasty or stenting procedures promotes vascular smooth muscle cell migration,23) proliferation24-26) and extracellular matrix production,27) with the intent to block this receptor by angiotensin-converting enzyme inhibitors or specific angiotensin II type 1 receptor antagonists and thereby reduce restenosis.28)29)
In the present study, ramiprilat-coated stents did not show marked anti-proliferative effects compared to control bare-metal stent, as reported previously using other drug-eluting stents, such as sirolimus-, paclitaxel-, or zotarolimus-eluting stents. However, ramiprilat-coated stents had good anti-inflammatory effects and promoted arterial healing compared to control bare-metal stents.
There were several limitations in the current study. First, it remains to be established whether or not our findings in normal, non-atherosclerotic porcine coronary arteries stimulated with oversized stents for neointimal proliferation can be extrapolated to human clinical situations with pre-existing atherosclerosis and stent sizes matched to the reference vessel. Second, our study did not examine the early inflammatory response, as there is little neointimal formation at that time. Third, we examined inflammation based on hematoxylin and eosin histologic findings. It would be more appropriate and accurate to utilize immunohistochemistry techniques, which are the standard for such studies, and with appropriate techniques can be used on resin-embedded sections. Fourth, we did not perform long-term follow-up experiments to examine late neointimal reactions, late inflammatory responses, and arterial healing. Fifth, we only used a single dose of ramiprilat for stent coating. In the future, a study using higher doses of ramiprilat coating onto the stent struts is needed.
In conclusion, although ramiprilat-coated stents did not show significant inhibitory effects with respect to neointimal hyperplasia, there were good effects on inflammatory reactions and arterial healing similar to control stents in a porcine coronary restenosis model.





 XML Download
XML Download